Nove publikacije
Nove ugotovitve prispevajo k boljšemu razumevanju vzrokov Rettovega sindroma
Zadnji pregled: 02.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.

Rettov sindrom je redka nevrološka razvojna motnja, za katero trenutno ni zdravila ali dobrega zdravljenja. Povzroča hude telesne in kognitivne simptome, od katerih se mnogi prekrivajo z motnjami avtističnega spektra.
Rettov sindrom povzročajo mutacije v genu MECP2, ki je močno izražen v možganih in ima očitno pomembno vlogo pri ohranjanju zdravja nevronov. Gen se nahaja na kromosomu X, sindrom pa prizadene predvsem dekleta. Za razvoj zdravljenja Rettovega sindroma želijo raziskovalci bolje razumeti MECP2 in njegove funkcije v možganih.
Raziskovalci, vključno s soustanoviteljem inštituta Whitehead Rudolfom Jaenischom, že desetletja preučujejo MECP2, vendar je veliko osnovnih dejstev o genu ostalo neznanih. Beljakovina, ki jo kodira gen MECP2, sodeluje pri regulaciji genov; veže se na DNK in vpliva na raven izražanja različnih drugih genov ali na količino beljakovin, ki jih ti proizvajajo.
Vendar pa raziskovalci niso imeli popolnega seznama genov, na katere vpliva MECP2, in ni bilo soglasja o tem, kako MECP2 vpliva na te gene.
Zgodnje študije MECP2 so nakazovale, da je represor, ki zmanjšuje izražanje ciljnih genov, vendar so raziskave Jaenischa in drugih že prej pokazale, da MECP2 deluje tudi kot aktivator, ki povečuje izražanje svojih tarč – in da je morda sploh aktivator. Prav tako ni znan mehanizem delovanja MECP2 oziroma kaj natančno protein počne, da povzroči spremembe v izražanju genov.
Zaradi omejitev tehnologije raziskovalcem ni uspelo dobiti jasnejših odgovorov na ta vprašanja. Vendar so Yanish, podoktorski raziskovalec njegovega laboratorija Yi Liu in Yanishev nekdanji član laboratorija Anthony Flamier, zdaj docent na raziskovalnem centru CHU Sainte-Justine na Univerzi v Montrealu, s pomočjo najsodobnejših tehnik odgovorili na ta preostala vprašanja o MECP2 in pridobili nove vpoglede v njegovo vlogo pri zdravju in boleznih možganov.
Njihovi rezultati so bili objavljeni v reviji Neuron, raziskovalci pa so ustvarili tudi spletni repozitorij svojih podatkov MECP2, portal MECP2-NeuroAtlas, kot vir za druge raziskovalce.
"Mislim, da bo ta članek temeljito spremenil razumevanje ljudi o tem, kako MECP2 povzroča Rettov sindrom. Imamo povsem novo razumevanje mehanizma in lahko ponudi nove možnosti za razvoj zdravljenja bolezni," pravi Janisch, ki je tudi profesor biologije na MIT.
Globlje razumevanje MECP2 v možganih
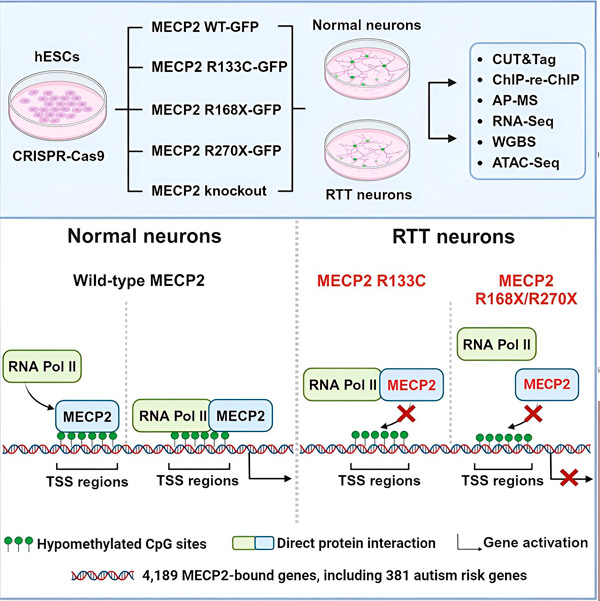
Raziskovalci so najprej ustvarili podroben zemljevid, kje se MECP2 veže v zaporedjih človeških nevronskih genov, bodisi znotraj genov bodisi v regulatornih regijah DNK v njihovi bližini. Uporabili so pristop, imenovan CUT&Tag, ki lahko z visoko natančnostjo določi interakcije beljakovin z DNK.
Raziskovalci so odkrili več kot 4000 genov, povezanih z MECP2. Kartiranje so ponovili v nevronih s pogostimi mutacijami MECP2, povezanimi z Rettovim sindromom, da bi ugotovili, kje je MECP2 v bolezenskem stanju izčrpan.
Poznavanje genov, na katere se MECP2 veže, je Liuju in Flamierju omogočilo, da sta začela povezovati tarče MECP2 z zdravjem možganov. Ugotovila sta, da so številne tarče vključene v razvoj in delovanje nevronskih aksonov in sinaps.
Prav tako so primerjali svoj seznam tarč MECP2 z bazo podatkov o genih, povezanih z avtizmom, Simons Foundation Autism Research Initiative (SFARI) in ugotovili, da je 381 genov v tej bazi podatkov tarč MECP2.
Vir: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Te ugotovitve lahko pomagajo razjasniti mehanizme, ki so podlaga za simptome avtizma pri Rettovem sindromu, in predstavljajo dobro izhodišče za raziskovanje možne vloge MECP2 pri avtizmu.
»Ustvarili smo prvi integriran zemljevid epigenoma MECP2 v zdravju in bolezni, ta zemljevid pa lahko vodi prihodnje raziskave,« pravi Liu. »Poznavanje, kateri geni so tarče MECP2 in kateri geni so neposredno moteni pri bolezni, zagotavlja trdno podlago za razumevanje Rettovega sindroma in postavljanje vprašanj o regulaciji genov v nevronih.«
Raziskovalci so preučevali tudi, ali MECP2 povečuje ali zmanjšuje izražanje svojih ciljnih genov. V skladu z zgodovino, ko so nekateri MECP2 identificirali kot aktivator, drugi pa kot represor, sta Liu in Flamier našla primere, kjer je MECP2 igral obe vlogi.
Čeprav MECP2 pogosteje velja za represor, sta Liu in Flamier ugotovila, da je večinoma aktivator – kar potrjuje prejšnje ugotovitve Jaenischa in Liuja. En nov poskus je pokazal, da MECP2 aktivira vsaj 80 % svojih tarč, drug pa, da aktivira do 88 % svojih tarč.
Zemljevid ciljnih genov, ki so ga ustvarili raziskovalci, je ponudil dodaten vpogled v vlogo MECP2 kot aktivatorja. Ugotovili so, da se geni, ki jih MECP2 aktivira, običajno vežejo na območje DNK pred genom, imenovano mesto začetka transkripcije.
To je mesto, kjer celični mehanizem sproži proces prepisovanja gena v RNA, nakar se RNA prevede v funkcionalni protein, ki je produkt izražanja genov. Prisotnost MECP2 na mestu začetka transkripcije, kjer se začne izražanje genov, je skladna z njegovo vlogo aktivatorja genov.
Raziskovalci so se nato lotili ugotavljanja, kakšno vlogo ima MECP2 pri aktivaciji genov. Preučili so, na katere molekule se MECP2 veže na tem mestu, poleg DNK, in ugotovili, da MECP2 neposredno interagira z beljakovinskim kompleksom, imenovanim RNA polimeraza II (RNA Pol II). RNA Pol II je ključni celični stroj, ki prepisuje DNK v RNA. RNA Pol II sama ne more najti genov, zato potrebuje različne kofaktorje oziroma beljakovinske sodelavce, ki ji pomagajo pri opravljanju njenega dela.
Raziskovalci predlagajo, da MECP2 služi kot eden takšnih kofaktorjev, ki pomaga RNA Pol II sprožiti transkripcijo na genih, kjer se MECP2 veže. Strukturna analiza MECP2 je identificirala dele molekule, ki se vežejo na RNA Pol II, drugi poskusi pa so potrdili, da izguba MECP2 zmanjša prisotnost RNA Pol II na ustreznih mestih začetka transkripcije, kot tudi raven izražanja ciljnih genov.
To nakazuje, da je Rettov sindrom lahko posledica zmanjšane transkripcije genov, na katere cilja MECP2, zaradi mutacij MECP2, ki preprečujejo vezavo na RNA Pol II ali vezavo na DNK. V skladu s to idejo so najpogostejše mutacije MECP2, povezane z boleznijo, okrnjene: mutacije, pri katerih manjka del proteina, kar lahko spremeni interakcijo med MECP2 in RNA Pol II.
Raziskovalci upajo, da njihove ugotovitve ne bodo le spremenile našega razumevanja MECP2, temveč da bi globlje in širše razumevanje vpliva MECP2 na razvoj in delovanje možganov lahko privedlo do novih spoznanj, ki bodo pomagala ljudem z Rettovim sindromom in sorodnimi motnjami, vključno z avtizmom.
»Ta projekt je odličen primer sodelovalne narave Janischevega laboratorija,« pravi Flamier. »Z Rudolfom sva imela specifično težavo, povezano z Rettovim sindromom, jaz pa sem imel izkušnje s tehnologijo CUT&Tag, ki bi lahko rešila težavo. Skozi razpravo sva ugotovila, da lahko združiva svoja prizadevanja in zdaj imava odlično zbirko informacij o MECP2 in njegovih povezavah z boleznijo.«