Medicinski strokovnjak članka

Nove publikacije
Mukopolisaharidoza tipa 3
Zadnji pregled: 04.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
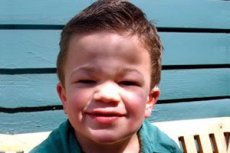
Mukopolisaharidoza tipa III (sinonimi: Sanfilippov sindrom, pomanjkanje lizosomske α-acetilglukozaminidaze - mukopolisaharidoza III A, pomanjkanje acetil-CoA-α-glukozaminid-N-acetiltransferaze - mukopolisaharidoza III B, N-acetilglukozamin-6-sulfataza - mukopolisaharidoza III C, pomanjkanje sulfamidaze - mukopolisaharidoza III D).
 [
[ Patogeneza
Bolezen povzročajo mutacije v štirih različnih genih: lizosomski α-acetilglukozaminidazi (mukopolisaharidoza III A), acetil-CoA-α-glukozaminid-N-acetiltransferazi (mukopolisaharidoza III B), lizosomski N-acetilglukozamin-6-sulfatazi (mukopolisaharidoza III C) in sulfamidazi (mukopolisaharidoza III D). Vsi encimi sodelujejo pri presnovi heparan sulfata.
Gen za heparan-N-sulfatazo – SGSH – se nahaja na dolgem kraku kromosoma 17 – 17q25.3. 75,3 % trenutno znanih mutacij v genu SGSH so točkovne mutacije. Opisane so bile pogosto pojavljajoče se mutacije, značilne za evropske populacije – R74C (56 % na Poljskem in 21 % v Nemčiji) in R245H (56 % na Nizozemskem).
Pogostost mutacije R74C je 47,5 %, mutacije R245H pa 7,5 %. Drugi dve opisani mutaciji, delll35G in N389S, skupaj predstavljata 21,7 % mutiranih alelov.
Gen za aN-acetil-glukozaminidazo (NAGLU) se nahaja na dolgem kraku kromosoma 17 - 17q21. 69 % mutacij, najdenih v genu NAGLU, so missense in nesmiselne mutacije, 26,3 % pa so majhne delecije in insercije. Gen za acetil-CoA-cc-glukozaminid-N-acetiltransferazo (HGSNAT) se nahaja na kratkem kraku kromosoma 8 - 8p11.1. Gen je bil karakteriziran šele leta 2006 in do danes je bilo v njem najdenih le nekaj mutacij.
Gen za N-acetil-glukozamin-6-sulfatazo - GNS - se nahaja na dolgem kraku kromosoma 12 - 12ql4. V svetu je registriranih 12 bolnikov z mukopolisaharidozo IIID. Opisane so štiri mutacije v genu GNS.
Pri vseh podtipih mukopolisaharidoze III pride do motnje v razgradnji heparan sulfata, ki je del strukture celičnih membran, vključno z nevronskimi membranami, kar je povezano s hudim nevrodegenerativnim procesom, ki ga povzroča kortikalna atrofija. Kronična driska je pojasnjena z vključenostjo avtonomnega živčnega sistema v patološki proces skupaj z disfunkcijo črevesne sluznice. Senzorinevralna izguba sluha je verjetno posledica treh vzrokov: pogostega otitisa, deformacije slušnih koščic in anomalij notranjega ušesa. Okornost sklepov je posledica deformacije metafiz, odebelitev sklepne kapsule pa je sekundarna zaradi odlaganja glikozaminoglikanov in fibroze. Intrasindromske razlike v resnosti bolezni so izključno posledica preostale funkcionalne aktivnosti mutantnega encima: višja kot je, blažja je bolezen.
Simptomi mukopolisaharidoza tipa 3
Klinični polimorfizem pri Sanfilippovem sindromu je manj izrazit kot pri drugih vrstah mukopolisaharidoze. Značilno je počasno napredovanje bolezni, hude nevrološke motnje z blagimi simptomi iz notranjih organov in drugih sistemov.
Prvi simptomi bolezni se običajno pojavijo med 2. in 6. letom starosti pri otrocih s prej normalnim razvojem. Manifestni simptomi vključujejo nazadovanje psihomotoričnega in govornega razvoja, psihiatrične motnje v obliki sindroma hiperaktivnosti, avtistično ali agresivno vedenje, motnje spanja; otroci postanejo neprevidni in nepozorni.
Drugi pogosti simptomi so hirzutizem, groba dlaka, zmerna hepatosplenomegalija, valgusna deformacija okončin in kratek vrat. Razvoj grobih obraznih potez, kot je gargojilizem, in skeletnih deformacij, kot je multipla dizostoza, so pri mukopolisaharidozi III šibko izraženi v primerjavi z drugimi vrstami mukopolisaharidoze, za katere je značilen Hurlerjev fenotip. Višina praviloma ustreza starosti, okorelost sklepov pa redko povzroča disfunkcijo. Večina bolnikov pogosto razvije osteoporozo in osteomalacijo. Sekundarne skeletne motnje - veliko tveganje za patološke zlome. Hude psihonevrološke motnje se najpogosteje opazijo v 6. do 10. letu življenja in vodijo do izrazite socialne neprilagojenosti. Progresivna senzorinevralna izguba sluha je lastna vsem bolnikom s hudimi in zmernimi oblikami bolezni. Konvulzije se pojavijo pri skoraj vseh bolnikih, ko bolezen napreduje.
Bolezen hitro napreduje in večina bolnikov ne preživi 20. leta starosti. Mukopolisaharidoza IIIA velja za najpogostejšo in najhujšo vrsto tega sindroma.
Diagnostika mukopolisaharidoza tipa 3
Diagnozo mukopolisaharidoze III potrdimo z določanjem ravni izločanja glikozaminoglikanov v urinu in merjenjem aktivnosti encimov. V primeru mukopolisaharidoze III se poveča skupno izločanje glikozaminoglikanov v urinu in opazimo hiperizločanje heparan sulfata. Aktivnost lizosomskih encimov, ki ustrezajo določenemu podtipu mukopolisaharidoze III, merimo v levkocitih ali kulturi kožnih fibroblastov z uporabo umetnega fluorogenega substrata.
Prenatalna diagnostika je mogoča z merjenjem encimske aktivnosti v biopsiji horionskih resic v 9.–11. tednu nosečnosti in/ali določanjem spektra glikozaminoglikanov v amnijski tekočini v 20.–22. tednu nosečnosti. Pri družinah z znanim genotipom se lahko DNK diagnostika izvede v zgodnji nosečnosti.
Katere teste so potrebne?
Diferencialna diagnoza
Diferencialna diagnostika se izvaja tako znotraj skupine mukopolisaharidoz kot tudi z drugimi boleznimi lizosomskega shranjevanja: mukolipidozami, galaktosialidozo, sialidozo, manozidozo, fukozidozo, gangliozidozo GM1.
Koga se lahko obrnete?
Zdravljenje mukopolisaharidoza tipa 3
Do danes niso bile razvite učinkovite metode zdravljenja mukopolisaharidoze III. Indicirano je simptomatsko zdravljenje.
Использованная литература