Medicinski strokovnjak članka

Nove publikacije
Usherjev sindrom
Zadnji pregled: 04.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Usherjev sindrom je dedna bolezen, ki se kaže kot popolna gluhost od rojstva, pa tudi kot progresivna slepota s starostjo. Izguba vida je povezana s pigmentnim retinitisom, procesom pigmentne degeneracije mrežnice. Mnogi ljudje z Usherjevim sindromom imajo tudi hude težave z ravnotežjem.
Epidemiologija
Zahvaljujoč raziskavi je bilo mogoče ugotoviti, da Usherjev sindrom prizadene približno 8 % pregledanih gluhonemih otrok (testi so bili opravljeni v posebnih ustanovah za gluhoneme osebe). Pigmentni retinitis so opazili pri 6–10 % bolnikov s prirojeno gluhostjo, ki jo posledično opazimo pri približno 30 % ljudi s pigmentno boleznijo mrežnice.
Menijo, da se ta bolezen po vsem svetu pojavlja pri približno 3–10 ljudeh od 100 tisoč. Pojavi se lahko enako pri ženskah in moških. Približno 5–6 % svetovnega prebivalstva trpi za tem sindromom. Približno 10 % vseh primerov otroške hude gluhosti je posledica Usherjevega sindroma I. in II. tipa.
V Združenih državah Amerike sta najpogostejša tipa 1 in 2. Skupaj predstavljata približno 90 do 95 odstotkov vseh primerov Usherjevega sindroma pri otrocih.
Vzroki Usherjev sindrom
Usherjev sindrom tipov I, II in III ima avtosomno recesivni vzrok, medtem ko tip IV velja za motnjo kromosoma X. Vzroki za slepoto in gluhost, ki se pojavljata pri tem sindromu, še niso dovolj raziskani. Domneva se, da so ljudje s to boleznijo preobčutljivi na komponente, ki lahko poškodujejo strukturo DNK. Poleg tega je ta bolezen lahko povezana z motnjami imunskega sistema, vendar v tem primeru ni natančne slike tega procesa.
Leta 1989 so bile kromosomske nepravilnosti prvič odkrite pri bolnikih z boleznijo tipa II, kar bi lahko v prihodnosti privedlo do načina izolacije genov, ki povzročajo sindrom. Morda bi bilo mogoče te gene identificirati tudi pri nosilcih in razviti posebne prenatalne genetske teste.
 [ 8 ]
[ 8 ]
Dejavniki tveganja
Sindrom se deduje, ko sta prizadeta oba starša, tj. deduje se po recesivnem tipu. Otrok lahko podeduje bolezen tudi, če sta njegova starša nosilca gena. Če imata oba bodoča starša ta gen, je verjetnost, da bo imel otroka s tem sindromom, 1 proti 4. Oseba, ki ima samo en gen za sindrom, velja za nosilca, vendar nima simptomov motnje. Dandanes še ni mogoče ugotoviti, ali ima oseba gen za to bolezen.
Če se otrok rodi staršem, od katerih eden nima takega gena, je verjetnost, da bo podedoval sindrom, zelo majhna, vendar bo zagotovo nosilec.
Simptomi Usherjev sindrom
Simptomi Usherjevega sindroma vključujejo izgubo sluha in nenormalno kopičenje pigmentiranih celic v očesnih strukturah. Bolnik nato razvije degeneracijo mrežnice, ki povzroči poslabšanje vida in v najhujših primerih končno izgubo vida.
Senzorinevralna izguba sluha je lahko blaga ali popolna in se običajno ne razvije od rojstva. Vendar pa se lahko pigmentna bolezen mrežnice začne razvijati v otroštvu ali kasneje. Rezultati testov so pokazali, da se lahko centralna ostrina vida ohrani več let, tudi ko se periferni vid poslabša (stanje, imenovano "tunelski vid").
To so glavne manifestacije bolezni, ki jih lahko včasih dopolnijo še druge motnje, kot so psihoza in druge duševne motnje, težave z notranjim ušesom in/ali siva mrena.
Obrazci
Med raziskavo so bile ugotovljene 3 vrste te bolezni, pa tudi 4. oblika, ki je precej redka.
Za tip I bolezni je značilna prirojena popolna gluhost, pa tudi motnja ravnotežja. Pogosto taki otroci začnejo hoditi šele pri starosti 1,5 leta. Poslabšanje vida se običajno začne pri 10 letih, končni razvoj stanja nočne slepote pa se začne pri 20 letih. Otroci s to vrsto bolezni lahko razvijejo progresivno poslabšanje perifernega vida.
Pri bolezni tipa II opazimo zmerno ali prirojeno gluhost. V tem primeru se poslabšanje delne gluhosti pogosto ne pojavi več. Pigmentni retinitis se začne razvijati okoli konca adolescence ali po 20. letu. Razvoj nočne slepote se običajno začne pri 29–31 letih. Okvara vidne ostrine pri patologiji tipa II običajno napreduje nekoliko počasneje kot pri tipu I.
Za III. tip bolezni je značilna progresivna izguba sluha, ki se običajno začne med puberteto, pa tudi postopen razvoj retinitisa pigmentosa v istem obdobju (nekoliko kasneje kot izguba sluha), ki lahko postane dejavnik pri razvoju progresivne slepote.
Manifestacije patologije tipa IV se pojavljajo predvsem pri moških. V tem primeru opazimo tudi progresivne motnje ter izgubo sluha in vida. Ta oblika je zelo redka in ima običajno X-kromosomsko naravo.
Diagnostika Usherjev sindrom
Diagnoza Usherjevega sindroma se postavi na podlagi bolnikove opažene kombinacije nenadne gluhosti in progresivne izgube vida.
Testi
Za odkrivanje mutacije se lahko naroči poseben genetski test.
Ugotovljenih je bilo enajst genetskih lokusov, ki lahko povzročijo razvoj Usherjevega sindroma, in devet genov, ki so zagotovo vzrok za to motnjo:
- Tip 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tip 2: ush2a, VLGR1, WHRN.
- Usherjev sindrom tipa 3: USH3A.
Znanstveniki NIDCD so skupaj s kolegi z univerz v New Yorku in Izraelu odkrili mutacijo, imenovano R245X, v genu Pcdh15, ki predstavlja velik odstotek Usherjevega sindroma tipa 1 v judovski populaciji.
Če želite izvedeti več o laboratorijih, ki izvajajo klinična preskušanja, obiščite https://www.genetests.org in v imeniku laboratorijev poiščite »Usherjev sindrom«.
Če želite izvedeti več o obstoječih kliničnih preskušanjih, ki vključujejo genetsko testiranje za Usherjev sindrom, obiščite https://www.clinicaltrials.gov in poiščite »Usherjev sindrom« ali »genetsko testiranje za Usherjev sindrom«.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentalna diagnostika
Obstaja več metod instrumentalne diagnostike:
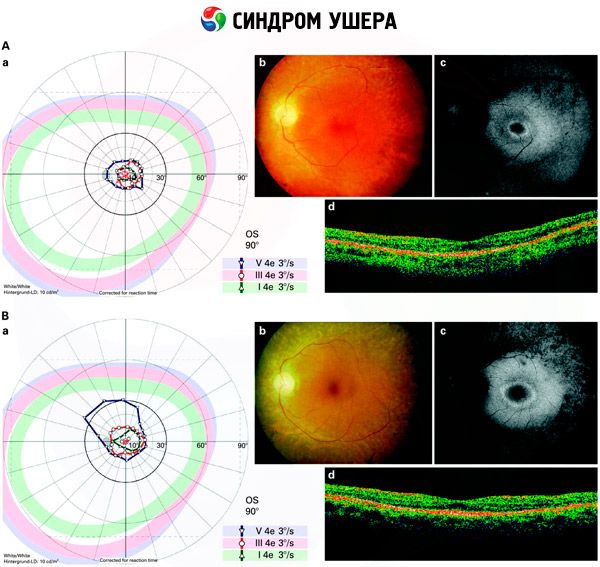
- Pregled fundusa za odkrivanje prisotnosti pigmentnih madežev na mrežnici, kot tudi zožitev mrežničnih žil;
- Elektroretinogram, ki omogoča odkrivanje začetnih degenerativnih odstopanj v očesni mrežnici. Prikazuje izumrtje elektroradiografskih poti;
- Elektronistagmogram (ENG) meri nehotene gibe oči, ki bi lahko kazali na prisotnost neravnovesja.
- Avdiometrija, ki se uporablja za ugotavljanje prisotnosti gluhosti in njene stopnje.
Diferencialna diagnoza
Usherjev sindrom je treba razlikovati od nekaterih podobnih motenj.
Hallgrenov sindrom, za katerega je značilna prirojena izguba sluha in progresivna izguba vida (razvijeta se tudi katarakta in nistagmus). Dodatni simptomi vključujejo ataksijo, psihomotorične motnje, psihozo in duševno zaostalost.
Alstromov sindrom, dedna bolezen, pri kateri mrežnica degenerira, kar povzroči izgubo centralnega vida. Ta sindrom je povezan z debelostjo pri otrocih. Hkrati se po 10 letih začneta razvijati sladkorna bolezen in izguba sluha.
Rubela pri nosečnici v prvem trimesečju lahko povzroči različne nepravilnosti v razvoju otroka. Med posledicami takšne nepravilnosti so izguba sluha, pa tudi (ali) težave z vidom, poleg tega pa še različne razvojne napake.
Koga se lahko obrnete?
Zdravljenje Usherjev sindrom
Trenutno ni zdravila za Usherjev sindrom. Zato terapija v tem primeru vključuje predvsem upočasnitev procesa izgube vida in kompenzacijo izgube sluha. Možne metode zdravljenja vključujejo:
- Jemanje vitamina A (nekateri oftalmologi menijo, da lahko visoki odmerki palmitata vitamina A upočasnijo, ne pa ustavijo napredovanja pigmentnega retinitisa);
- Vstavitev posebnih elektronskih naprav v pacientova ušesa (slušni aparati, kohlearni vsadki).
Oftalmologi priporočajo, da večina odraslih s pogostimi oblikami retinitisa pigmentosa dnevno jemlje 15.000 ie (mednarodnih enot) vitamina A palmitata pod nadzorom. Ker ljudje z Usherjevim sindromom tipa 1 niso bili vključeni v študijo, visoki odmerki vitamina A za to skupino bolnikov niso priporočljivi. Ljudje, ki razmišljajo o jemanju vitamina A, se morajo o tej možnosti zdravljenja pogovoriti s svojim zdravnikom. Druga priporočila za to možnost zdravljenja vključujejo:
- Spremenite svojo prehrano, da vključite živila z visoko vsebnostjo vitamina A.
- Ženske, ki načrtujejo nosečnost, naj prenehajo jemati visoke odmerke vitamina A tri mesece pred načrtovano zanositvijo zaradi povečanega tveganja za prirojene okvare.
- Nosečnice naj prenehajo jemati visoke odmerke vitamina A zaradi povečanega tveganja za prirojene okvare.
Pomembno je tudi, da takšnega otroka prilagodimo družabnemu življenju. To zahteva pomoč specialnih pedagogov in psihologov. V primeru, da je bolnik začel postopoma izgubljati vid, ga je treba naučiti uporabljati znakovni jezik.
Napoved
Usherjev sindrom ima neugodno prognozo. Pri večini bolnikov s to boleznijo katere koli vrste se vidno polje in njegova ostrina začneta slabšati v obdobju 20-30 let. V nekaterih primerih pride do popolne dvostranske izgube vida. Izguba sluha, ki jo vedno spremlja nemota, se zelo hitro razvije v popolno dvostransko izgubo sluha.