Medicinski strokovnjak članka

Nove publikacije
Sindrom Aper
Zadnji pregled: 04.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.

 [
[ Vzroki Sindrom Aper
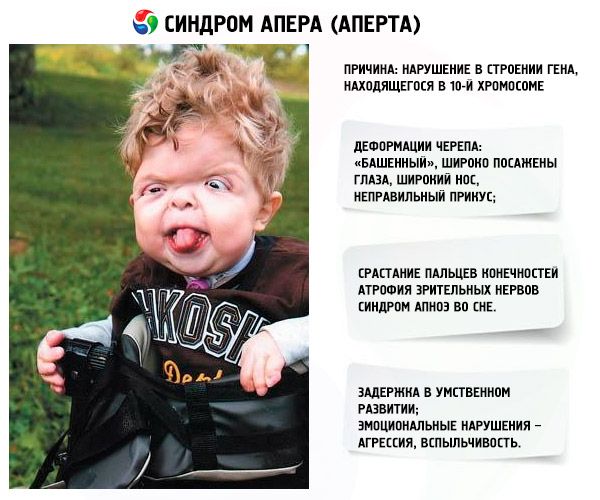
Razvoj sindroma izzove motnja v strukturi gena, ki se nahaja na kromosomu 10. Ta gen je odgovoren za proces ločevanja prstov in poleg tega za pravočasno zapiranje lobanjskih šivov.
Poleg tega se za vzroke patologije štejejo nalezljive bolezni, prebolene med nosečnostjo (kot so rdečke ali tuberkuloza, sifilis ali gripa, pa tudi meningitis), in izpostavljenost matere rentgenskim žarkom. Najpogosteje se tak sindrom odkrije pri otrocih, rojenih starejšim staršem.
Patogeneza
Apertov sindrom je deden. Njegov tip je avtosomno dominanten (tj. v družini, v kateri je eden od bodočih staršev bolan, je verjetnost, da bo imel otrok to bolezen, 50–100 %).
Edinstvena mutacija v receptorju 2 za rastni faktor fibroblastov (FGFR2) povzroči povečano število progenitorskih celic, ki se razvijejo vzdolž osteogene poti. To na koncu povzroči povečano subperiostalno tvorbo kostnega matriksa in prezgodnjo osifikacijo lobanjskih šivov med fetalnim razvojem. Vrstni red in hitrost taljenja šivov določata stopnjo deformacije in invalidnosti. Ko se šivalni material zaceli, je rast drugih tkiv, pravokotnih na ta šiv, omejena, zraščene kosti pa delujejo kot enoten kostni oder.
Prvi genetski dokaz, da je sindaktilija pri Apertovem sindromu posledica okvare receptorja za rastni faktor keratinocitov (KGFR), je bilo opažanje korelacije med izražanjem KGFR v fibroblastih in resnostjo sindaktilije.
Ambliopija in strabizem sta pogostejša pri bolnikih z mutacijo FGFR2 Ser252Trp, atrofija optičnega diska pa je pogostejša pri bolnikih z mutacijo FGFR2 Pro253Arg. Bolniki z mutacijami FGR2 Ser252Trp imajo bistveno večjo prevalenco okvare vida v primerjavi z bolniki z mutacijo FGFR2 Pro253Arg.
Simptomi Sindrom Aper
Nekatere manifestacije bolezni so jasno opazne že ob rojstvu otroka, saj se razvijejo v maternici. Med glavnimi simptomi sindroma so:
- Lobanja je deformirana - razteza se v višino in pridobi videz "stolpa"; poleg tega so oči široko postavljene in rahlo izbočene (ker se velikost očesnih jamic zmanjša); nos se razširi in oblikuje se napačen ugriz (zgornji zobje prekomerno štrlijo);
- Prsti okončin so popolnoma zraščeni (predvsem prstanec, pa tudi sredinec in kazalec) in so videti kot kožna membrana ali popolna fuzija kosti; poleg tega lahko zrastejo dodatni prsti;
- Duševna zaostalost (ne pojavlja se pri vseh);
- Optični živci atrofirajo, zaradi česar se ostrina vida zmanjša (v nekaterih primerih se razvije popolna izguba vida);
- Povišan intrakranialni tlak, ki nastane kot posledica prezgodnjega zaprtja lobanjskih šivov, se kaže v obliki glavobolov in bruhanja s slabostjo;
- Ker zgornja čeljust ostaja nerazvita, se pojavijo težave z dihanjem;
- Sindrom spalne apneje je pogost pojav.
- Čustvene manifestacije – agresija, pomanjkanje zadržanosti, močna razdražljivost.
Zapleti in posledice
Diagnostika Sindrom Aper
Za postavitev diagnoze so potrebni naslednji koraki:
- Zdravnik mora analizirati bolnikove pritožbe in zdravstveno anamnezo. Ugotoviti je treba, ali so bili v družini primeri podobne patologije;
- Nevrološki pregled za oceno oblike lobanje in intelektualnega razvoja pacienta (posebni vprašalniki in intervju);
- Pregled fundusa za odkrivanje prisotnosti simptomov povečanega intrakranialnega tlaka (otekanje optičnega diska in zamegljenost njegovih robov);
- Za oceno stanja lobanje se opravi rentgensko slikanje;
- Računalniško in magnetno resonančno slikanje glave za pregled strukture možganov in lobanje plast za plastjo, za ugotavljanje prisotnosti simptomov prezgodnjega zraščanja lobanjskih šivov in poleg tega hidrocefalusa (zaradi povečanega intrakranialnega tlaka se kopiči odvečna cerebrospinalna tekočina (to je cerebrospinalna tekočina, ki spodbuja presnovni proces in prehrano možganov));
- Rentgensko slikanje stopal in rok za ugotavljanje vzroka zlitja prstov (to je pomembno za načrtovanje nadaljnjega kirurškega posega);
- Morda bo predpisan posvet z medicinskim genetikom in nevrokirurgom.
Testi
Izvaja se genetska analiza pogostih mutacij, ki se pojavljajo v genu tipa FGFR2.
Diferencialna diagnoza
Ta sindrom je treba razlikovati od drugih genetskih patologij, pri katerih se opazi kraniosinostoza. To so bolezni, kot so Pfeifferjev, Crouzonov, Saethre-Chotzenov in Carpenterjev sindrom. Za izključitev teh anomalij se uporabljajo molekularno-genetske metode testiranja.
Koga se lahko obrnete?
Zdravljenje Sindrom Aper
Kirurgija velja za edino učinkovito zdravljenje Apertovega sindroma – pomaga odpraviti individualne telesne napake in tudi duševno zaostalost.
Med tem postopkom se koronalni šiv zapre, da se prepreči morebitna poškodba možganov. Najpogostejša tehnika je kraniofacialna distrakcija, ki vključuje stopnjevano vleko lobanje. Ortodontska in/ali ortognatska kirurgija se izvaja za odstranitev posameznih obraznih napak.
Poleg tega se bolnikom izvaja kirurška korekcija fuzije prstov.
Reference
Medicinska genetika. Nacionalni vodnik. Uredil Ginter EK, Puzyrev VP GEOTAR-Media. 2022