Medicinski strokovnjak članka

Nove publikacije
Keratoderma: vzroki, simptomi, diagnoza, zdravljenje
Zadnji pregled: 07.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
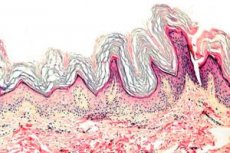
Keratoderma je skupina dermatoz, za katere je značilna motena proces keratinizacije - prekomerno nastajanje poroženele kože predvsem na dlaneh in podplatih.
Vzroki in patogeneza bolezni niso povsem pojasnjeni. Raziskave so pokazale, da keratoderme povzročajo mutacije v genih, ki kodirajo keratin 6, 9, 16. Pomanjkanje vitamina A, hormonske disfunkcije, predvsem spolnih žlez, ter bakterijske in virusne okužbe imajo velik pomen pri patogenezi. So eden od simptomov dednih bolezni in tumorjev notranjih organov (parapsoriatične keratoderme).
Simptomi. Razlikujemo med difuzno (Unna-Tostova keratoderma, Meleda keratoderma, Papillon-Lefevreova keratoderma, mutilirajoča keratoderma in sindromi, ki vključujejo difuzno keratodermo kot enega glavnih simptomov) in fokalno (diseminirana pikčasta keratoderma Fischer-Buschke, akrokeratoelastoidoza Kosti, omejena keratoderma Bruhauer-Franzesthesti, linearna keratoderma Fuchsa itd.) keratodermo.
Winy-Tostova keratoderma (sinonimi: prirojena ihtioza dlani in podplatov, Winy-Tostov sindrom) se prenaša avtosomno dominantno. Prisotna je difuzna prekomerna keratinizacija kože dlani in podplatov (včasih samo podplatov), ki se razvije v prvih dveh letih življenja. Patološki proces na koži se začne z rahlim odebeljevanjem kože dlani in podplatov v obliki traku eritema lividne barve na meji z zdravo kožo. Sčasoma se na njihovi površini pojavijo gladke, rumenkaste poroženele plasti. Lezija se redko razširi na hrbtno stran zapestij ali prstov. Pri nekaterih bolnikih se lahko pojavijo površinske ali globoke razpoke, opazimo pa tudi lokalno hiperhidrozo. Pri bolniku, ki ga je opazoval avtor, so za Winy-Tostovo keratodermo trpeli stric po materini strani, brat in sin.
Opisani so primeri poškodbe nohtov (odebelitev), zob in las pri keratodermi Winy-Tost v kombinaciji z različnimi anomalijami skeleta in patologijami notranjih organov, živčnega in endokrinega sistema.
Histopatologija. Histološki pregled razkrije izrazito hiperkeratozo, granulozo, akantozo in majhne vnetne infiltrate v zgornjem dermisu. Diferencialna diagnoza. Bolezen je treba razlikovati od drugih vrst keratoderme.
Meledina keratoderma (sinonimi: Meledina bolezen, prirojeni progresivni akrokeratom, Siemensova palmoplantarna transgradientna keratoza, Kogoyeva dedna palmoplantarna progresivna keratoza) se deduje avtosomno recesivno. Za to obliko keratoderme so značilne debele, rumeno-rjave poroženele plasti z globokimi razpokami. Ob robovih lezije je viden vijolično-vijoličen rob, širok nekaj milimetrov. Proces se običajno razširi na zadnji del rok in stopal, podlakti in goleni. Večina bolnikov ima lokalno hiperhidrozo. V zvezi s tem postane površina dlani in podplatov rahlo vlažna in prekrita s črnimi pikami (izvodi znojnih žlez).
Bolezen se lahko razvije do 15. do 20. leta starosti. Nohti se odebelijo in deformirajo.
Histopatologija. Histološki pregled razkrije hiperkeratozo, včasih akantozo in kronični vnetni infiltrat v papilarnem dermisu.
Diferencialna diagnoza. Melelno keratodermo je treba razlikovati od Unna-Tostove keratoderme.
Keratoderma Papillon-Lefevre (sinonim: palmoplantarna hiperkeratoza s parodontitisom) se deduje avtosomno recesivno.
Bolezen se kaže v 2.-3. letu življenja. Klinična slika bolezni je podobna Melelini bolezni. Poleg tega so značilne spremembe na zobeh (nenormalnosti pri izraščanju mlečnih in stalnih zob z razvojem kariesa, gingivitisa, hitro napredujoče parodontalne bolezni s prezgodnjo izgubo zob).
Histopatologija. Histološki pregled razkrije odebelitev vseh plasti povrhnjice, zlasti poroženele plasti, in neznatne celične skupke limfocitov in histiocitov v dermisu.
Diferencialna diagnoza. Bolezen je treba razlikovati od drugih keratoderm. Pomembna razlikovalna značilnost je značilna zobna patologija, ki je pri drugih oblikah dednih difuznih keratoderm ne najdemo.
Keratoderma mutilans (sinonimi: Fonwinkelov sindrom, dedni mutilirajoči keratom) je vrsta difuzne keratoderme, ki se deduje avtosomno dominantno. Razvije se v drugem letu življenja in je značilna po difuznih poroženelih oblogah na koži dlani in podplatov s hiperhidrozo. Sčasoma se na prstih oblikujejo vrvičasti žlebovi, kar vodi do kontraktur in spontane amputacije prstov. Folikularna keratoza se izraža na hrbtni strani rok, pa tudi v predelu komolčnih in kolenskih sklepov. Nohtne plošče so spremenjene (pogosto kot urna stekla). Opisani so primeri hipogonadizma, rubinaste alopecije, izgube sluha, pahionihije.
Histopatologija. Histološki pregled razkrije hudo hiperkeratozo, granulozo, akantozo in majhne vnetne infiltrate v dermisu, ki jih sestavljajo limfociti in histiociti.
Diferencialna diagnoza. Pri razlikovanju mutilirajoče keratoderme od drugih oblik difuzne keratoderme je treba najprej upoštevati učinek mutilacije, ki ni značilen za druge oblike. Pri diferencialni diagnozi vseh oblik difuzne keratoderme je treba vedeti, da je lahko eden glavnih simptomov številnih dednih sindromov.
Zdravljenje. Neotigazon je indiciran pri splošni terapiji keratoderme. Odmerek zdravila je odvisen od resnosti procesa in znaša 0,3–1 mg/kg bolnikove teže. V odsotnosti neotigazona se priporoča vitamin A v odmerku od 100 do 300.000 mg na dan dlje časa. Zunanja terapija obsega uporabo mazil z aromatičnimi retinoidi, keratolitiki in steroidnimi sredstvi.
 [
[ Kaj te moti?
Kaj je treba preveriti?
Kako preučiti?