Medicinski strokovnjak članka

Nove publikacije
Alkaptonurija je prirojena encimska nepravilnost
Zadnji pregled: 04.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Ena zelo redkih presnovnih motenj, alkaptonurija, se nanaša na prirojene anomalije v presnovi aminokisline tirozin.
Ta sindrom se lahko imenuje tudi pomanjkanje homogentisat oksidaze, homogentisinurija, dedna ohronoza ali bolezen črnega urina.[ 1 ]
Epidemiologija
Po statističnih podatkih ni več kot devet primerov alkaptonurije na milijon ljudi. V večini evropskih držav pa je en primer na 100–250 tisoč živorojenih otrok.
Med evropskimi državami je izjema Slovaška (zlasti relativno majhna severozahodna regija), kjer je prevalenca alkaptonurije en primer na 19.000 novorojenčkov. To je najverjetneje posledica dejstva, da je med slovaškimi romskimi družinami, ki tam živijo, stopnja inbridinga (poroke med bratranci in sestričnami) najvišja v Evropi: 10–14 %. [ 2 ]
Vzroki alkaptonurija
Natančni vzroki za alkaptonurijo kot prirojeno motnjo katabolizma (presnovne razgradnje) aromatske (homociklične) α-aminokisline tirozina so bili ugotovljeni: ta vrsta presnovne motnje je posledica homozigotnih ali sestavljenih heterozigotnih mutacij enega od tisočih genov na kromosomu 3, natančneje gena HGD na lokusu 3q21-q23 na dolgem kraku kromosoma. Ta gen kodira nukleotidna zaporedja jetrnega encima homogentisat-1,2-dioksigenaze [ 3 ] (imenovane tudi homogentizinska kislinska oksidaza ali homogentisat oksidaza) - metaloproteina, ki vsebuje železo in je potreben za eno od stopenj razgradnje tirozina v telesu. [ 4 ], [ 5 ]
Alkaptonurija je torej okvara encima homogentisat-1,2-dioksigenaze oziroma natančneje posledica njegove genetsko pogojene pomanjkljivosti ali popolne odsotnosti. [ 6 ]
Ker gre za prirojeno pomanjkanje encimov, se alkaptonurija deduje avtosomno recesivno, kar pomeni, da morata za pojav alkaptonurije pri otrocih oba starša imeti spremenjen gen za encim, saj vsak od njiju otroku prenese le eno kopijo gena od obeh razpoložljivih.
Po najnovejših podatkih obstaja več kot dvesto variant modifikacije gena HGD, najpogosteje pa opazimo missense mutacije, translokacijo in spajanje.
Dejavniki tveganja
Edini dejavnik tveganja za razvoj te prirojene encimopatije je njena prisotnost v družinski anamnezi in dedovanje dveh spremenjenih kopij gena HGD, če starši nimajo alkaptonurije (tveganje za prenos anomalije je 25 %) ali če ima eden od staršev to motnjo. [ 7 ]
Patogeneza
Tirozin igra ključno vlogo pri sintezi beljakovin, proizvodnji kromoproteinov – kožnega pigmenta melanina, pa tudi ščitničnih hormonov in kateholaminskih nevrotransmiterjev.
Mehanizem za uravnavanje količine tirozina v celicah je zelo zapleten, telo pa normalizira njegovo presežno vsebnost z razgradnjo. Proces katabolizma tirozina je, tako kot vse aromatske aminokisline, večstopenjski in poteka v več fazah. Vsaka faza presnovne razgradnje tirozina poteka s sodelovanjem specifičnega encima in nastankom vmesne spojine.
Torej, najprej se aminokislina razgradi v para-hidroksifenilpiruvat, ki se pretvori v alkapton – 2,5-dihidroksifenilocetno ali homogentizinsko kislino. Nato bi se moral alkapton pretvoriti v maleocetno kislino, vendar se to ne zgodi. [ 8 ]
In patogeneza alkaptonurije je sestavljena iz prenehanja biokemijskih reakcij katabolizma tirozina v fazi tvorbe homogentizinske kisline: za njeno razgradnjo preprosto ni potreben encim – homogentizat oksidaza.
Homogentizinske kisline telo ne uporablja in se lahko kopiči z izločanjem skozi ledvice. Poleg tega se oksidira v benzokinoacetat (benzokinonoocetna kislina), ki z vezavo na molekule tkiv in telesnih tekočin tvori biopolimerne spojine, obarvane kot melanin.
Kopičenje teh vmesnih produktov v tkivu vodi do motenj kolagene strukture hrustančnega tkiva, kar zmanjša njegovo elastičnost – s pojavom številnih kliničnih znakov alkaptonurije in razvojem zapletov.
Simptomi alkaptonurija
Za alkaptonurijo pri novorojenčkih in dojenčkih je značilno potemnenje urina. Ko je urin na plenicah, plenicah in spodnjem perilu izpostavljen zraku, postane temno rjav; to je posledica kopičenja in sproščanja homogentizinske kisline, ki se oksidira v benzokinoacetat. [ 9 ]
Če ni drugih simptomov, alkaptonurije pri majhnih otrocih pogosto ne prepoznamo pravočasno, saj lahko urin po več urah uriniranja potemni. Po nekaterih podatkih je v kliničnih okoljih odkrita le petina otrok, mlajših od 12 mesecev, ki so se rodili s pomanjkanjem tega encima. Zato je zelo pomembno, da starši pozorno spremljajo svoje dojenčke.
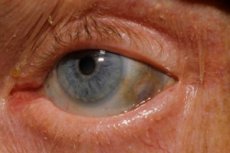
Poleg tega so med zgodnje znakedi pigmentacija (modrikasto-siva barva) beločnice oči in hrustanca ušes in nosu, kar pogosto imenujemo ohronoza.[ 10 ]
Sčasoma se pojavijo drugi simptomi:
- huda pigmentacija kože na ličnicah, pazduhah in genitalijah;
- obarvanje oblačil ob stiku z znojnimi deli telesa;
- napadi splošne šibkosti;
- hripav glas.
Upoštevati je treba, da sta alkaptonurija in ohronoza, kot je navedeno zgoraj, sinonimna imena za isto motnjo katabolizma tirozina.
Bolezen javorjevega urina in alkaptonurija. Prirojena bolezen javorjevega urina ali levcinoza je prav tako presnovna motnja, ima enak vzorec dedovanja in celo mutacije se pojavljajo na istem kromosomu, vendar vplivajo na gen, ki kodira encimski kompleks razvejane verige α-ketokisline dehidrogenaze. Zaradi tega telo ne more razgraditi nekaterih komponent beljakovin, zlasti aminokislin levcin, izolevcin in valin. Pri tej bolezni ima urin (in ušesno maslo) sladek vonj; poleg tega klinična slika te vrste organske acidemije vključuje hipopigmentacijo, nihanje krvnega tlaka, epileptične napade, bruhanje in drisko, padec ravni glukoze v krvi, ketoacidozo, halucinacije itd. Stopnja umrljivosti pri otrocih je precej visoka; pri odraslih lahko brez zdravljenja pride do kome in smrti zaradi možganskega edema.
Albinizem in alkaptonurija sta »združena« le s tirozinom. Albinizem, vključno z okulokutanim, povzročajo genetske mutacije, ki vplivajo na proizvodnjo pigmenta melanina. Prirojene spremembe so opažene v genu TYR na kromosomu 11 (11q14.3), ki kodira tirozinazo, melanosomski encim, ki vsebuje baker in je potreben za tvorbo kožnega pigmenta na osnovi produktov presnove tirozina. Ta bolezen je veliko pogostejša kot alkaptonurija.
Zapleti in posledice
Posledice in zapleti alkaptonurije, ki jo povzroča delovanje vmesnih presnovkov tirozina – homogentizinske in benzokinonocetne kisline – se pojavijo zaradi odlaganja reaktivnih pigmentiranih polimerov, uničenja kolagenskih fibril in poslabšanja stanja hrustanca (z zmanjšanjem njihove odpornosti na mehanske obremenitve).
Z leti se v odrasli dobi razvijeta degenerativni artritis in osteoartritis velikih sklepov (kolka, križno-iliakalnega in kolenskega sklepa); medvretenčni prostori se zožijo (zlasti v ledveni in prsni hrbtenici) – s kalcifikacijo in nastankom osteofitov; gostota tkiva subhondralnih kostnih plošč se zmanjša, spodaj ležeče kosti pa se lahko patološko preoblikujejo z nastankom izrastkov in deformacij. [ 11 ]
Zaradi iste kalcifikacije se lahko opazijo poškodbe srčnih zaklopk (aortne in mitralne) in koronarnih arterij – z znaki koronarne srčne bolezni, pa tudi nastanek kamnov v ledvicah in prostati. [ 12 ], [ 13 ]
Diagnostika alkaptonurija
Običajno diagnoza prirojenih presnovnih motenj temelji na preučevanju bioloških tekočin telesa.
Na podlagi katerih testov in reakcij lahko diagnosticiramo alkaptonurijo? Za odkrivanje homogentizinske kisline in določitev njene ravni (normalna – 20–30 mg na dan, povišana – 3–8 g) so potrebni testi urina. Vzorec urina se pregleda s plinsko kromatografijo ali masno spektrometrijo, pri čemer se uporabi tekočinska kromatografija; možen je presejalni test za prisotnost železovega klorida v urinu. [ 14 ]
Obstaja tudi metoda za hitro diagnostiko – določanje alkaptona v posušenih madežih urina na papirju (po intenzivnosti barve).
Pri razjasnitvi diagnoze instrumentalna diagnostika (radiografija) vključuje prepoznavanje radioloških znakov osteoartritisa in drugih sklepnih patologij pri bolnikih.
Diagnozo potrdijo molekularno genetske metode diagnosticiranja dednih bolezni, kot sta genetsko testiranje in sekvenciranje DNK. [ 15 ]
Diferencialna diagnoza
Diferencialna diagnoza vključuje hemokromatozo in akutno odpoved jeter pri novorojenčku, melanurijo, akutno intermitentno porfirijo, hemofagocitno limfohistiocitozo, primarno mitohondrijsko patologijo, revmatoidni artritis, ankilozirajoči spondilitis.
Koga se lahko obrnete?
Zdravljenje alkaptonurija
Glavno zdravljenje alkaptonurije je peroralno dajanje velikih odmerkov (vsaj 1000 mg na dan) askorbinske kisline. Pri otrocih to poveča izločanje homogentizinske kisline z urinom, pri odraslih pa zmanjša vsebnost njenega derivata, benzokinonocetne kisline, v urinu in upočasni njeno vezavo na vezivno tkivo sklepov in kolagena. [ 16 ]
Zahodnoevropske klinike testirajo zdravilo Nitizinon (Orfalin), zdravilo iz skupine metabolitov, ki zavira drugo stopnjo katabolizma tirozina: pretvorbo para-hidroksifenilpiruvata v homogentizinsko kislino. Vendar pa uporaba tega farmakološkega sredstva vodi do kopičenja tirozina in lahko povzroči hude stranske učinke, vključno z motnostjo roženice in fotofobijo, krvavitvami iz nosu in želodca, odpovedjo jeter, spremembami v krvi itd. Kljub temu je v Združenih državah Amerike Nitizinon odobren s strani FDA za zdravljenje tirozinemije tipa I. [17 ], [ 18 ]
Zato se pri težavah s sklepi, ki jih povzroča alkaptonurija, izvaja fizioterapija – vadbena terapija za povečanje mišične moči in izboljšanje gibljivosti sklepov ter balneoterapija in peloidna terapija za zmanjšanje bolečine.
Čeprav tirozin ne vnašamo le s hrano, ampak ga telo tudi proizvaja, se bolnikom z alkaptonurijo priporoča dieta z nizko vsebnostjo beljakovin in omejitev uživanja živil, bogatih s tirozinom, predvsem govedine in svinjine, mlečnih izdelkov (zlasti sirov), stročnic, oreščkov in semen.
Preprečevanje
Preprečevanje genskih mutacij je nemogoče, vendar za preprečevanje rojstva otrok z visokim tveganjem za prirojene motnje obstaja medicinsko genetsko svetovanje, ki je pred načrtovano nosečnostjo potrebno za tiste pare, katerih družinska anamneza vključuje dedne bolezni. [ 19 ]
Napoved
Smrtni izidi zaradi alkaptonurije so zelo redki, smrt pa lahko povzročijo resni zapleti, ki vključujejo srce in ledvice. Zato je splošna pričakovana življenjska doba ljudi z alkaptonurijo dobra.
Vendar pa se kakovost življenja zmanjša zaradi intenzivnih bolečin v sklepih ali hrbtenici z znatno omejitvijo gibljivosti, ki pogosto napredujejo.