Medicinski strokovnjak članka

Nove publikacije
Angelmanov sindrom pri otrocih in odraslih
Zadnji pregled: 04.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.

Obstaja vrsta bolezni, pri katerih se izrazi, kot je "pazi nase in ne boš zbolel", slišijo vsaj smešno. To so patologije, pri katerih so nekatere duševne in telesne nepravilnosti prirojene otrokovemu telesu že pred rojstvom, vendar starši za to niso krivi. Takšne bolezni povzročajo mutacije ali nepravilnosti v kromosomskih sklopih in se imenujejo kromosomske ali genetske. Angelmanov sindrom, Downov sindrom, Patauov sindrom, Edwardsov sindrom, Turnerjev sindrom, Prader-Willijev sindrom - to je le del genetskih bolezni z dokaj spodobnega seznama.
Sindrom srečnega človeka
Tokrat bomo govorili o patologiji, poimenovani po angleškem pediatru Harryju Angelmanu, ki je prvi sprožil to vprašanje leta 1965, potem ko je dan prej v svoji praksi srečal tri nenavadne otroke, ki so jih družili skupni nenavadni simptomi. Zdravnik je te otroke poimenoval otroci-lutke in o njih napisal članek, ki se je sprva imenoval "Otroci-marionete". Sam članek in njegov naslov sta bila napisana pod vtisom slike, ki so jo videli v enem od muzejev v Veroni. Slika je upodabljala smejočega se dečka in se je imenovala "Lutkovni deček". Povezava otroka, upodobljenega na sliki, s tremi otroki, ki jih je Angelman nekoč srečal v svoji praksi, je pediatra spodbudila, da je otroke zaradi bolezni, ki so jo imeli, združil v eno skupino.
Nič presenetljivega ni v tem, da otrok, omenjenih v članku, drugi zdravniki niso opazili. Navsezadnje se je na prvi pogled zdelo, da imajo popolnoma različne bolezni, tako različna je bila splošna klinična slika bolezni v treh različnih primerih. Morda bi "nova" kromosomska patologija zanimala tudi druge znanstvenike, vendar genetika takrat še ni bila dovolj razvita, da bi potrdila hipotezo angleškega zdravnika. Zato je bil članek po določenem zanimanju zanj za dolgo časa vržen na zadnjo polico.
Naslednja omemba Angelmanovega sindroma, kot se je zdaj imenoval članek angleškega pediatra G. Angelmana, sega v zgodnja 80. leta 20. stoletja. In šele leta 1987 je bilo mogoče najti razlog, zakaj se majhen del otrok rodi s takšnimi odstopanji, da se od zunaj zdijo nenehno nasmejani in srečni. Pravzaprav to sploh ni res, nasmeh pa je le grimasa, za katero se skriva nesrečna človeška duša in bolečina staršev.
Epidemiologija
Po statističnih podatkih se lahko kromosomska mutacija pri otroku razvije tako v ozadju podobnih mutacij pri starših kot tudi v odsotnosti le-teh. Ni jasne dedne narave Angelmanovega sindroma (AS), vendar je verjetnost razvoja patologije pri starših s kromosomskimi mutacijami precej visoka.
Zanimivo je tudi, da če ima družina že otroka z AS, obstaja enoodstotna možnost, da se bo rodil drugi otrok z isto motnjo, tudi če sta starša zdrava.
Še vedno ni natančnih statističnih podatkov o številu bolnikov z Angelmanovim sindromom. Morda je razlog v raznolikosti simptomov, ki se lahko pojavijo v določeni sestavi ali pa se dolgo časa sploh ne pojavijo. Domneva se, da je razširjenost bolezni: 1 otrok na 20.000 novorojenčkov. Vendar je ta številka zelo približna.
Vzroki Angelmanov sindrom
Angelmanov sindrom je medicinsko ime za kromosomsko patologijo, vendar še zdaleč ni edino. Ljudje to bolezen imenujejo sindrom otrok punčk, sindrom srečne lutke, Petruškin sindrom in sindrom smejoče se lutke. Ljudje si izmišljujejo najrazličnejša imena (včasih celo žaljiva za same bolnike in njihove starše), vendar je bolezen bolezen, ne glede na to, kako smešna je videti in ne glede na razloge.
In razlogi za razvoj Angelmanovega sindroma, tako kot mnoge druge genetske patologije, so v vseh primerih motnje v strukturi enega od kromosomov ali kromosomskega nabora kot celote. Toda v našem primeru je celoten problem v kromosomu 15, ki se je prenesel od matere. To pomeni, da očetov kromosom v tem primeru nima odstopanj, ženski pa je podvržen določenim mutacijam.
Angelmanov sindrom je glede na vrsto kromosomske nepravilnosti razvrščen kot kromosomska mutacija. Takšne mutacije so:
- Delecija (odsotnost dela kromosoma, ki vsebuje določen nabor genov; če eden od genov manjka, govorimo o mikrodeleciji), ki je posledica dveh prelomov in ene ponovne združitve, ko se izgubi del prvotnega kromosoma.
- Podvajanje (prisotnost dodatnega dela v kromosomu, ki je kopija obstoječega), ki v večini primerov vodi do smrti osebe, redkeje pa do neplodnosti.
- Inverzija (obrat enega od odsekov kromosoma za 180 stopinj, torej v nasprotno smer, nato pa se geni v njem nahajajo v nasprotnem vrstnem redu), ko so prekinjeni konci kromosoma povezani v vrstnem redu, ki je drugačen od prvotnega.
- Vstavitev (če del genskega materiala v kromosomu ni na svojem mestu),
- translokacija (če je določen del kromosoma pritrjen na drug kromosom; takšna mutacija je lahko obojestranska brez izgube delov).
Če otrok od nič hudega sluteče matere prejme mutiran kromosom, je obsojen na rojstvo z nepravilnostmi. Najpogostejši vzrok Angelmanovega sindroma še vedno velja za delecijo materinega 15. kromosoma, ko manjka majhen del. Manj pogoste mutacije pri sindromu "smejoče se lutke" so:
- premestitev,
- unipaternalna disomija (če je otrok od očeta prejel par kromosomov, materin kromosom manjka),
- mutacija genov v DNK, ki so tako glavni gradbeni (genetski) material kot tudi navodila za njegovo pravilno uporabo (zlasti mutacija gena ube3a v materinem kromosomu).
Prisotnost ene od teh mutacij pri starših je dejavnik tveganja za razvoj Angelmanovega sindroma pri otrocih. Vendar pa ne le kromosomske mutacije, temveč tudi genomske (ki so povezane s kvantitativno spremembo kromosomskih naborov in so pogostejše od kromosomskih) lahko izzovejo razvoj bolezni pri otroku. Med pogoste genomske mutacije spada kromosomska trisomija (če ima oseba v kromosomskem naboru več kot 46 kromosomov).
Za pojav patologije pri otroku sploh ni nujno, da imajo starši kromosomske nepravilnosti. Pa vendar obstaja določen odstotek bolnikov, pri katerih je bolezen dedna.
Patogeneza
Poglobimo se v biologijo oziroma natančneje v genetiko. Genetske informacije vsakega posameznega človeškega organizma so vsebovane v 23 parih kromosomov. En kromosom iz para se na otroka prenese od očeta, drugega pa od matere. Vsi pari kromosomov se razlikujejo po obliki in velikosti ter nosijo določene informacije. Tako je 23. par kromosomov (kromosomi X in Y) odgovoren za oblikovanje spolnih značilnosti otroka (XX - deklica, XY - fant, medtem ko lahko kromosom Y otrok prejme le od očeta).
V idealnem primeru otrok od staršev prejme 46 kromosomov, ki oblikujejo njegove genetske značilnosti in ga vnaprej določajo kot posameznika. Večje število kromosomov se imenuje trisomija in velja za odstopanje od norme. Na primer, prisotnost kromosoma 47 v kromosomskem naboru (kariotip, ki določa vrsto in individualne značilnosti) povzroči pojav Downovega sindroma.
Če kromosome obarvamo s posebnim barvilom, lahko pod mikroskopom vzdolž vsakega od njih vidimo proge različnih odtenkov. Znotraj vsake proge je ogromno število genov. Vse te proge so znanstveniki oštevilčili in imajo fiksno lokacijo. Odsotnost ene od prog se šteje za odstopanje od norme. Pri Angelmanovem sindromu lahko zelo pogosto opazimo odsotnost segmentov materinega kromosoma v intervalu q11-q13, ki se nahaja v dolgem kraku, katerega število baz DNK je le približno 4 milijone.
Glavna komponenta kromosoma velja za neverjetno dolgo molekulo DNK, ki vsebuje na tisoče genov in na desetine ter stotine milijonov dušikovih baz. Tako kromosom 15, odgovoren za razvoj Angelmanovega sindroma in številnih drugih, vsebuje 1200 genov in približno 100 milijonov baz. Vsaka motnja v strukturi molekule DNK bo zagotovo vplivala na videz in razvoj nerojenega otroka.
Genetske informacije, ki jih vsebujejo geni, se pretvorijo v beljakovine ali RNK. Ta proces se imenuje izražanje genov. Na ta način genetske informacije, prejete od staršev, dobijo tako obliko kot vsebino, ki se uteleša v njihovem edinstvenem ženskem ali moškem dediču.
Obstajajo številne patologije z neklasičnim tipom dedovanja, vključno z Angelmanovim sindromom, pri katerem geni, prejeti od staršev kot del parnih kromosomov, nosijo edinstven odtis staršev in se manifestirajo na različne načine.
Angelmanov sindrom je torej presenetljiv primer genomskega imprintinga, pri katerem je izražanje genov v otrokovem telesu neposredno odvisno od tega, od katerega starša so bili prejeti aleli (različne oblike enega gena, prejete od očeta in matere, ki se nahajajo na enakih odsekih parnih kromosomov). To pomeni, da le anomalije v materinem kromosomu vodijo do razvoja sindroma, medtem ko mutacije in strukturne motnje očetovega kromosoma povzročajo povsem drugačne patologije.
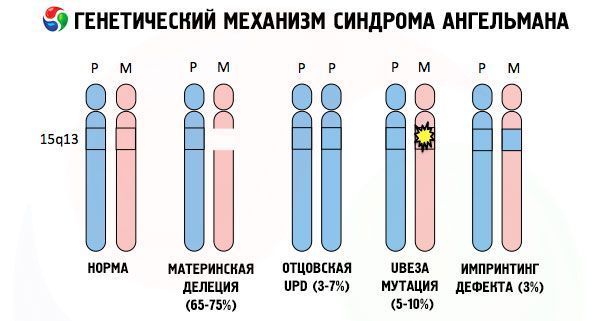
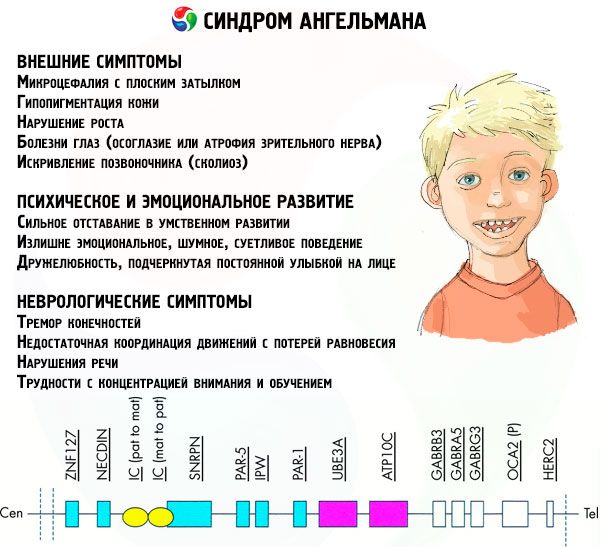
Pri tej patologiji pride do pomanjkanja določenih genov v materinem kromosomu ali do izgube/zmanjšanja aktivnosti posameznih genov (v veliki večini primerov gena ube3a, ki sodeluje pri presnovi ubikvitina, beljakovine, ki uravnava razgradnjo drugih beljakovin). Posledično se pri otroku diagnosticirajo duševne razvojne nepravilnosti in telesne deformacije.
Simptomi Angelmanov sindrom
Simptomi Angelmanovega sindroma vplivajo na različne vidike otrokovega življenja in razvoja: fizični, nevrološki, duševni. Na podlagi tega je mogoče prepoznati 3 skupine simptomov, ki kažejo na razvoj te patologije.
- Zunanji ali fizični simptomi:
- nesorazmerno majhna glava v primerjavi s telesom in okončinami, ki so normalne velikosti,
- preširoka usta,
- skoraj vedno je na obrazu nasmeh (z odprtimi usti),
- redki zobje,
- ozka zgornja ustnica,
- pogosto izbočen širok jezik,
- štrleča spodnja čeljust,
- koničasta brada,
- zelo svetla koža, pogosto lasje (albinizem, povezan z dejstvom, da telo ne proizvaja pigmenta melanina),
- temne lise na svetli koži (hipopigmentacija zaradi nezadostne proizvodnje melanina)
- fizični ali zunanji simptomi: očesne bolezni, kot sta strabizem ali atrofija vidnega živca,
- ukrivljenost hrbtenice (skolioza),
- toge noge (pri hoji oseba zaradi nizke gibljivosti sklepov ne upogne nog v kolenih, od tod tudi primerjava z lutkino hojo).
- Simptomi, povezani z duševnim in čustvenim razvojem:
- huda duševna zaostalost,
- pretirano čustveno, hrupno, sitno vedenje,
- pogosto ploskanje z rokami,
- izražena prijaznost, poudarjena s stalnim nasmehom na obrazu,
- pogost smeh brez razloga.
- Nevrološki simptomi:
- tremor okončin,
- nezadostna koordinacija gibov z izgubo ravnotežja,
- zmanjšan mišični tonus,
- različne motnje spanja,
- pogosti histerični napadi v otroštvu,
- motnje govora (otrok začne govoriti pozno, ima slabe komunikacijske sposobnosti in nerazločen govor),
- hiperaktivnost na ozadju povečane razdražljivosti,
- težave s koncentracijo in učenjem.
Vendar je to posplošena slika bolezni. Pravzaprav je klinična slika Angelmanovega sindroma v veliki meri odvisna od stopnje razvoja bolezni in vrste kromosomske mutacije, ki je povzročila patologijo. To pomeni, da se simptomi bolezni pri različnih bolnikih lahko bistveno razlikujejo, kar nam dolgo časa ni omogočalo razlikovanja patologije od drugih s podobno klinično sliko.
Med skupnim številom simptomov lahko izpostavimo tiste, ki so značilni za vse bolnike brez izjeme:
- huda duševna zaostalost,
- neprimerno vedenje (nerazumen smeh, povečana razdražljivost, slaba koncentracija, stanje evforije),
- nerazvitost motoričnih sposobnosti,
- slaba koordinacija gibov, ataksija hoje (neenakomeren tempo, zibanje z ene strani na drugo itd.), tresenje okončin.
- motnja govornega razvoja s prevlado neverbalnih komunikacijskih sredstev.
Med simptomi, s katerimi se srečuje velika večina bolnikov, lahko ločimo naslednje:
- nesorazmerje med glavo in telesom zaradi zaostalega telesnega razvoja,
- pri mnogih bolnikih je oblika lobanje takšna, da velikost možganov ostaja manjša kot pri zdravih ljudeh (mikrocefalija),
- epileptični napadi pred 3. letom starosti s progresivnim zmanjšanjem moči in pogostosti v starejši starosti,
- popačenje parametrov EEG (nihanja in visoka amplituda nizkofrekvenčnih valov).
Ti simptomi so precej pogosti, vendar jih 20 % bolnikov z Angelmanovim sindromom nima.
Še manj pogosto je mogoče diagnosticirati takšne manifestacije bolezni, kot so:
- hud ali blag strabizem,
- slab nadzor nad gibanjem jezika, zaradi česar bolniki pogosto brez razloga iztisnejo jezik,
- težave s požiranjem in sesanjem, zlasti pri majhnih otrocih,
- motnje pigmentacije kože in oči,
- roke dvignjene ali pokrčene med hojo,
- hiperrefleksija,
- motnje spanja, zlasti v otroštvu,
- pogosto slinjenje,
- nepotešljiva žeja,
- pretirano aktivni žvečilni gibi,
- preobčutljivost na vročino,
- raven zadnji del glave,
- štrleča spodnja čeljust,
- gladke dlani.
Precej velik odstotek bolnikov ima težave z uriniranjem, ki ga slabo nadzorujejo, oslabljene fine motorične sposobnosti, kar povzroča težave pri samooskrbi in učenju, ter prekomerno telesno težo. Skoraj vsi bolniki doživijo puberteto pozneje kot zdravi vrstniki.
Otroci z Angelmanovim sindromom dobro zaznavajo in razumejo ustni govor, vendar ne želijo sodelovati v pogovoru, saj svoj govor omejujejo na nekaj deset besed, potrebnih v vsakdanjem življenju. Vendar pa so takšni bolniki v odrasli dobi videti mlajši od svojih vrstnikov brez genetskih patologij.
Mnogi simptomi Angelmanovega sindroma so nestalni, zato se klinična slika bolezni s starostjo bistveno spreminja. Konvulzije in epileptični napadi postanejo manj pogosti ali popolnoma izginejo, bolnik postane manj razdražljiv in spanec se izboljša.
Zapleti in posledice
Angelmanov sindrom je huda, trenutno praktično neozdravljiva kromosomska patologija, ki bolnikom odvzema možnost normalnega življenja. Kakšno bo življenje otroka z AS, je v veliki meri odvisno od vrste kromosomske nepravilnosti.
Podvajanje kromosomskega segmenta je v večini primerov nezdružljivo z življenjem. In tudi če taki bolniki ne umrejo v otroštvu in dosežejo puberteto, nimajo možnosti, da bi imeli otroke.
Izbris ali odsotnost dela genov, ki se najpogosteje pojavlja pri Angelmanovem sindromu, je ovira za otrokovo učenje hoje in govora. Takšni otroci imajo hujšo obliko duševne zaostalosti, epileptični napadi pa se pojavljajo pogosteje, njihova intenzivnost pa je veliko večja kot pri bolnikih z drugimi kromosomskimi nepravilnostmi.
Če gre le za mutacijo enega gena, lahko otroka z ustrezno pozornostjo in pristopom naučimo osnov samooskrbe, komunikacije in interakcije v skupini, čeprav bo v razvoju še vedno zaostajal za vrstniki.
Za otroke z Angelmanovim sindromom, ki so po naravi prijazni, je najpomembnejša ljubezen in pozornost staršev. Le v tem primeru bo otrokovo izobraževanje obrodilo sadove, četudi majhne. Seveda se bolniki z AS ne bodo mogli učiti v redni šoli. Potrebujejo posebne razrede, kjer se bodo otroci najprej naučili koncentracije, nato pa bodo postopoma dobivali osnove šolskega znanja.
Diagnostika Angelmanov sindrom
Angelmanov sindrom je prirojena razvojna patologija. Vendar ga je zaradi določenih okoliščin pogosto nemogoče diagnosticirati v povojih in zgodnjem otroštvu. To je posledica nespecifičnosti in šibke izraženosti simptomov pri dojenčkih in otrocih, mlajših od 3 let. Razširjenost bolezni v naši državi ni tako velika, da bi jo zdravniki prepoznali med njenimi vrstniki.
Angelmanov sindrom pri dojenčkih se lahko kaže kot zmanjšan mišični tonus, kar se kaže v težavah s hranjenjem (šibkost sesalnega in požirajočega refleksa), kasneje pa v težavah pri učenju hoje (takšni otroci začnejo hoditi veliko kasneje). Ti simptomi so prvi znaki razvojne nepravilnosti pri dojenčku, ki je lahko povezana s kromosomsko nepravilnostjo. To domnevo lahko potrdi le genetska analiza.
Posebna pozornost je namenjena otrokom, katerih starši imajo različne genomske ali kromosomske motnje. Navsezadnje se bolezen sprva morda ne manifestira, in če se patologija odkrije pravočasno, je z začetkom intenzivnega dela z otrokom mogoče doseči bistveno večji uspeh pri učenju in upočasniti napredovanje bolezni.
Če imajo starši različne kromosomske nepravilnosti, se genetska analiza opravi še pred rojstvom otroka, saj je SA ena od patologij, ki jih je mogoče odkriti v embrionalni fazi.
Zbiranje materiala za genetske raziskave se lahko izvede na dva načina:
- invaziven (z določenim odstotkom tveganja, saj je za odvzem vzorca amnijske tekočine potrebno prodreti v maternico),
- neinvazivna (analiza otrokove DNK iz materine krvi).
Nato se izvedejo naslednje študije:
- fluorescentna in situ hibridizacija (metoda FISH) – vezava DNK sonde, označene s posebnim barvilom, na preučevano DNK, ki ji sledi pregled pod mikroskopom.
- analiza mutacij v genu ube3a in imprintiranih genih,
- Analiza metilacije DNK z uporabo posebnih metod, ki se uporabljajo v genetiki.
Genetski testi v primeru kromosomskih nepravilnosti zagotavljajo dokaj natančne informacije, kar pomeni, da bodoči starši vnaprej vedo, na kaj se morajo pripraviti. Vendar pa obstajajo izjeme. Pri določeni skupini bolnikov ob prisotnosti vseh simptomov, ki kažejo na patologijo, rezultati testov ostanejo normalni. To pomeni, da je patologijo mogoče prepoznati le s skrbnim opazovanjem otroka od zgodnjega otroštva: kako se prehranjuje, kdaj je začel hoditi in govoriti, ali pri hoji upogiba noge itd.
Poleg metode FISH lahko med instrumentalnimi diagnostičnimi metodami za Angelmanov sindrom izpostavimo tomografijo (CT ali MRI), ki pomaga določiti stanje in velikost možganov, ter elektroencefalogram (EEG), ki prikazuje delovanje posameznih delov možganov.
Zdravniki običajno postavijo končno diagnozo v starosti 3-7 let, ko ima bolnik že večino simptomov in je vidna dinamika razvoja bolezni.
Katere teste so potrebne?
Diferencialna diagnoza
Angelmanov sindrom je genetska patologija, ki praktično nima specifičnih manifestacij. Večina simptomov lahko enako kaže tako na AS kot na druge genetske patologije.
Diferencialna diagnoza Angelmanovega sindroma se izvaja z naslednjimi patologijami:
- Pitt-Hopkinsov sindrom (za bolnike je značilna duševna zaostalost, vesel značaj, nasmeh, imajo precej velika in široka usta, opazimo mikrocefalijo). Razlika je v napadih hiperventilacije in zadrževanju diha v budnem stanju.
- Christiansonov sindrom (bolniki so duševno zaostali ljudje z veselo naravo, ne morejo govoriti, za katere so značilni mikrocefalija, ataksija, konvulzije, nehotni gibi mišic).
- Mowat-Wilsonov sindrom (simptomi: duševna zaostalost, epileptični napadi, koničasta brada, odprta usta, srečen izraz na obrazu, mikrocefalija). Značilnosti: velika razdalja med očmi, oči poševno navznoter, zaobljena konica nosu, nazaj obrnjena ušesna školjka.
- Kabukijev sindrom (za katerega so značilni blaga do zmerna duševna zaostalost, težave z govorom in motoriko, mišična oslabelost, epileptični napadi, mikrocefalija, dolgi intervali med srbenjem in oslabljena koordinacija). Zanj so značilne obokane obrvi, obrnjen stranski del spodnje veke, široko postavljene oči, dolge palpebralne reže z dolgimi, gostimi trepalnicami.
- Rettov sindrom (diferenciacija od AS pri ženskah). Simptomi: zaostanek v razvoju govora, epileptični napadi, mikrocefalija. Razlika je v tem, da na obrazu ni veselega izraza, pojavljajo se napadi apneje in apraksije, ki sčasoma napredujejo.
- Sindrom avtosomno recesivnega duševnega zaostajanja 38 (simptomi: izrazita duševna zaostalost z zamudami v motoričnih sposobnostih in govoru, mišična oslabelost, težave s hranjenjem v otroštvu, impulzivnost). Razlikovalna značilnost je modra barva šarenice.
- Sindrom podvajanja gena MECP 2 (diferenciacija od SA pri moških). Simptomi: huda duševna zaostalost, mišična oslabelost od otroštva, težave z govorom ali pomanjkanje govora, epilepsija. Značilnosti: progresivna miopatija, nenehno ponavljajoče se okužbe.
- Kleefstrov sindrom (simptomi: težave z govorom in mišljenjem, mišična oslabelost, motnje spanja, pomanjkanje pozornosti, odprta usta, hiperaktivnost, epileptični napadi, ataksija, motnje ravnotežja). Značilnosti: raven obraz, kratek, pritlikav nos, široko postavljene oči, velika, obrnjena spodnja ustnica, agresivni izbruhi.
- Smith-Magenisov sindrom (za katerega so značilni epileptični napadi, težave s spanjem, motnje intelektualnega in motoričnega razvoja). Med značilne značilnosti spadata širok in raven obraz ter izrazito čelo.
- Koolen-de Vriesov sindrom (blaga do zmerna duševna zaostalost, mišična oslabelost, epileptični napadi, prijaznost). Razlikovalne značilnosti: dolg obraz z visokim čelom, štrleča ušesa, poševne oči, velika gibljivost sklepov, prirojene srčne napake.
- Phelan-McDermidov sindrom (simptomi: duševna zaostalost, motnje govora ali pomanjkanje govora). Značilnosti: velike roke z razvitimi mišicami, mišična oslabelost od rojstva, šibko potenje.
Takšne patologije, kot so pomanjkanje adenil sukcinata, sindrom avtosomno recesivnega duševnega razvoja 1, sindrom podvajanja kromosoma 2q23.1, sindromi haploinsuficience genov FOXG1, STXBP1 ali MEF2C in nekatere druge, se lahko "pohvalijo" s simptomi, podobnimi Angelmanovemu sindromu.
Naloga zdravnika je postaviti natančno diagnozo, razlikovati Angelmanov sindrom od patologij s podobnimi simptomi in predpisati učinkovito zdravljenje, ki je ustrezno diagnosticirani fazi bolezni.
Zdravljenje Angelmanov sindrom
Angelmanov sindrom je ena tistih patologij, za katere medicina še vedno išče učinkovito zdravljenje. Etiološko zdravljenje bolezni je v fazi razvoja različnih metod in sredstev, od katerih jih veliko še ni bilo preizkušenih na ljudeh. To pomeni, da se morajo zdravniki zaenkrat omejiti na simptomatsko terapijo, ki pomaga nekako ublažiti nezavidljiv položaj otrok in odraslih z marionetnim sindromom, ki trpijo zaradi epileptičnih napadov, slinjenja, hipotenzije in motenj spanja.
Tako je mogoče zmanjšati pogostost in moč epileptičnih napadov s pomočjo pravilno izbranega antikonvulzivnega zdravila. Toda celotna težava je v tem, da se napadi pri bolnikih s SA razlikujejo od običajnih epileptičnih napadov, saj jih zaznamuje več vrst napadov, kar pomeni, da je stanje mogoče ublažiti z dajanjem več zdravil hkrati.
Najbolj priljubljeni antikonvulzivi, ki se uporabljajo za zdravljenje Angelmanovega sindroma, so: valprojska kislina, topiramat, lamotrigin, levetiracetam, klonazepam in zdravila na njihovi osnovi. Manj pogosto se uporabljajo zdravila na osnovi karmazepina, fenitoina, fenobarbitala, etosuksimida, saj lahko nekatera od njih izzovejo paradoksalen učinek, ki se kaže v krepitvi in povečanju pogostosti epileptičnih napadov. To se zgodi, če se zdravilo uporablja kot del monoterapije.
Za zdravljenje slinjenja se običajno uporabljata dve metodi: medikamentozna (zdravila, ki zavirajo nastajanje sline) in kirurška, ki vključuje ponovno vsaditev slinčnih kanalov. Vendar pa se v primeru SA te metode štejejo za neučinkovite in vprašanje ostaja odprto. Starši in tisti, ki skrbijo za takšne bolnike, morajo temu vprašanju posvetiti posebno pozornost, saj bolniki sami običajno ne morejo nadzorovati slinjenja, nekateri pa preprosto niso sposobni poskrbeti zase.
Druga težava je kratko trajanje spanja. Otroci z Angelmanovim sindromom pogosto spijo največ 5 ur, kar negativno vpliva na delovanje celotnega telesa. Lahko vznemirljivi, aktivni otroci, ki imajo radi igre in komunikacijo (tudi če se poskušajo omejiti na neverbalne metode), so čez dan opazno utrujeni. Da bi se telo dobro spočilo, potrebuje globok, poln spanec, vendar je prav v tem ovira.
Zdi se, da bi morala biti pomirjujoča zdravila (fenotiazini in atipični antipsihotiki), ki pomirjajo živčni sistem, zadostna za izboljšanje spanca pri vznemirjenih bolnikih. Toda v primeru AS je uporaba takšnih zdravil polna negativnih učinkov. Zato zdravniki še vedno dajejo prednost blagim uspavalnim tabletam, kot sta melatonin (naravno hormonsko zdravilo na osnovi hormona spanja), ki se bolnikom daje uro pred spanjem v količini 1 tablete, in difenhidramin. Pogostost dajanja in odmerjanje določi zdravnik glede na stanje in starost bolnika.
Včasih imajo bolniki z Angelmanovim sindromom težave s prebavo in blatom. Blato lahko izboljšate z odvajali (po možnosti zeliščnimi).
Ali pa se lahko problema lotite drugače, kot so to storili ameriški zdravniki, ki temeljijo na nekaterih metodah zdravljenja avtizma, saj so številni simptomi, značilni za AS, značilni tudi za avtizem (impulzivnost, nehoteni gibi, ponavljajoča se dejanja, pomanjkanje pozornosti, težave s komunikacijo itd.). Ugotovljeno je bilo, da vnos hormona sekretina, ki normalizira prebavo in blato, pozitivno vpliva na pozornost bolnikov, oksitocin pa pomaga izboljšati otrokove kognitivne sposobnosti in spomin ter popraviti vedenje.
Res je, da sami hormoni niso dovolj, še posebej, ko gre za otroke. Pri Angelmanovem sindromu je indicirana vedenjska terapija, delo s psihologom in logopedom (poučevanje neverbalnih komunikacijskih metod in znakovnega jezika). Vzgoja takšnih otrok naj bi temeljila na individualnem programu s sodelovanjem posebej usposobljenih učiteljev, psihologa in staršev. Žal to ni povsod mogoče in družine ostanejo same s svojo težavo.
Ker veliko mladih bolnikov z AS trpi zaradi nizkega mišičnega tonusa in težav s sklepi, se veliko pozornosti posveča fizioterapiji. Najpogosteje se zdravniki zatečejo k uporabi parafinskih aplikacij, elektroforeze in magnetne terapije.
Aktivna tonična masaža in posebne vaje terapevtske telesne vzgoje bodo bolnemu otroku pomagale, da se čez nekaj časa postavi na noge in samozavestno hodi. V tem pogledu je še posebej koristna vodna gimnastika, ki jo priporočamo pri SA v hladni vodi. Poveča mišični tonus in otroka nauči nadzorovati svoje telo in usklajevati gibe.
Antikonvulzivno zdravljenje
Najnevarnejši simptom Angelmanovega sindroma so napadi, podobni tistim pri epilepsiji. Ta simptom opazimo pri 80 % bolnikov, kar pomeni, da je treba vsem predpisati učinkovito antikonvulzivno zdravljenje.
Zdravljenje epileptičnih napadov se izvaja s pomočjo vitaminov in antikonvulzivov. Pri Angelmanovem sindromu, ki ga spremlja konvulzivni sindrom, bodo koristni vitamini skupine B, pa tudi vitamini C, D in E. Vendar je samostojno predpisovanje vitaminske terapije v tem primeru zelo nevarno, saj lahko nenadzorovan vnos vitaminov zmanjša učinkovitost antiepileptikov in izzove nove, hujše in dolgotrajnejše napade.
Izbor antikonvulzivnih zdravil in predpisovanje njihovega učinkovitega odmerka naj bi opravil zdravnik specialist. Odloči tudi, ali bo eno zdravilo dovolj ali bo moral bolnik jemati dve ali več zdravil dlje časa.
Večini bolnikov zdravniki predpišejo zdravila z valprojsko kislino (Valprojska kislina, Depakine, Convulex, Valparin itd.), ki preprečujejo epileptične napade in izboljšujejo razpoloženje in duševno stanje bolnikov.
Valprojska kislina je na voljo v obliki tablet, sirupa in injekcijskih raztopin. Najbolj priljubljeno zdravilo je zdravilo s podaljšanim sproščanjem "Depakine" v tabletah in kot raztopina za intravensko dajanje. Odmerek zdravila določi zdravnik individualno, odvisno od teže, starosti in stanja bolnika.
Zdravilo se jemlje med obroki 2 do 3-krat na dan. Povprečni dnevni odmerek je 20-30 mg na 1 kilogram bolnikove teže, največji pa 50 mg/kg na dan.
Kontraindikacije za uporabo. Ne uporabljajte v primeru disfunkcije jeter in trebušne slinavke, hemoragične diateze, hepatitisa, porfirije in preobčutljivosti na zdravilo.
Neželeni učinki vključujejo tresenje rok, prebavne in blatne motnje ter spremembe telesne teže.
"Topiramat" je tudi zdravilo izbire za SA. Proizvaja se v obliki tablet in se uporablja tako kot del monoterapije kot v kombinaciji z drugimi zdravili.
Način uporabe in odmerjanje. Tablete jemljite peroralno ne glede na vnos hrane. Začetni dnevni odmerek za odrasle je 25–50 mg, za otroke pa 0,5–1 mg/kg. Vsak teden se odmerek poveča v skladu z zdravnikovimi navodili.
Zdravila se ne sme jemati med nosečnostjo in dojenjem, pa tudi v primeru preobčutljivosti na njegove sestavine. Zdravilo ima veliko različnih stranskih učinkov.
Zdravila, ki jih zdravnik lahko predpiše za Angelmanov sindrom: klomazepam, rivotril, lamotrigin, seizar, lamictal, levetiracetam, keppra, epiterra itd.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradicionalna medicina in homeopatija
Tradicionalna medicina, tako kot homeopatski pripravki, je seveda relativno varna, vendar je učinkovitost takšnega zdravljenja Angelmanovega sindroma lahko sporna.
Čeprav lahko ljudsko zdravljenje pri nekaterih stvareh še vedno pomaga. Govorimo o zaustavitvi epileptičnih napadov. V zvezi s tem je lahko zdravljenje z zelišči precej učinkovito.
Dober učinek zagotavlja zdravilna zbirka na osnovi potonike, sladkega korena in vodne leče (sestavine se vzamejo v enakih količinah). Zelišča je treba zmleti v moko. Po 2 tednih od začetka jemanja lahko opazite znatno zmanjšanje pogostosti napadov.
Pri krčih je koristen tudi sivkin prevretek (1 čajna žlička na kozarec vrele vode). Mešanico kuhamo 5 minut in pustimo stati pol ure. Zdravilo jemljemo ponoči 14 dni.
Vodna (ali alkoholna) infuzija maternice velja za učinkovito pri epileptičnih napadih.
Od homeopatskih pripravkov za preprečevanje napadov pri Angelmanovem sindromu lahko uporabite zdravila na osnovi kamilice in maternice, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Vendar je treba upoštevati, da lahko le homeopatski zdravnik predpiše učinkovite in varne odmerke zdravil v vsakem posameznem primeru.
Preprečevanje
Kot je bralec verjetno že razumel, medicina še ne more preprečiti genskih mutacij in drugih kromosomskih nepravilnosti, pa tudi popraviti situacije. To se lahko zgodi vsakomur, saj se otroci z Angelmanovim sindromom rodijo zdravim staršem, genetika, ki je trenutno ena najmanj raziskanih vej medicine, pa tega še ne more pojasniti.
Edino, kar lahko storimo, je, da se odgovorno lotimo načrtovanja nosečnosti, se pravočasno registriramo in opravimo preglede. A spet bo takšen ukrep bolj izobraževalen kot preventivan, tako kot vsi pregledi. Toda mladi starši bodo vnaprej vedeli, na kaj se morajo pripraviti, in v primeru pozitivnega odgovora se bodo odločili, ali lahko prevzamejo takšno odgovornost, kot je vzgoja bolnega otroka.
Napoved
Prognoza za Angelmanov sindrom je odvisna od narave kromosomske nepravilnosti in pravočasnosti njenega odkrivanja. Najbolj prizadeti so tisti otroci, katerih kromosom 15 vsebuje "vrzeli" v genih (delecija). Verjetnost, da bodo taki bolniki hodili in govorili, je izjemno nizka. Druge primere je mogoče popraviti s skrbnim pristopom in ljubeznijo do svojega otroka.
Žal takšni bolniki ne bodo mogli postati polnopravni člani družbe, kljub temu da še zdaleč niso neumni, razumejo govor in njegov pomen. Vendar bodo imeli težave s komunikacijo do konca življenja. Bolnike je mogoče znakovnega jezika učiti že od otroštva, vendar jih ni mogoče prisiliti, da komunicirajo z besedami. Besedišče "govorečih" bolnikov je omejeno na minimum besed, ki se uporabljajo v vsakdanjem življenju (5-15 besed).
Kar zadeva pričakovano življenjsko dobo in splošno zdravstveno stanje bolnikov z Angelmanovim sindromom, se številke tukaj gibljejo okoli povprečnih vrednosti. V odrasli dobi se bolniki večinoma soočajo z zdravstvenimi težavami, kot sta skolioza in debelost, ki pa ob pravilnem pristopu k zdravljenju niso smrtno nevarne.