Medicinski strokovnjak članka

Nove publikacije
Cistična fibroza
Zadnji pregled: 04.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Cistična fibroza je genetska avtosomno recesivna monogenska bolezen, za katero je značilna motnja izločanja eksokrinih žlez vitalnih organov s poškodbo predvsem dihal in prebavil, hudim potekom in neugodno prognozo.
 [ 1 ]
[ 1 ]
Epidemiologija
Incidenca cistične fibroze niha med 1:2500 in 1:4600 novorojenčkov. Vsako leto se po vsem svetu rodi približno 45.000 ljudi s cistično fibrozo. Incidenca nosilcev gena cistične fibroze je 3-4 %, pri čemer je po vsem svetu približno 275 milijonov ljudi nosilcev tega gena, od tega približno 5 milijonov v Rusiji in približno 12,5 milijona v državah SND.
Vzroki cistična fibroza
Cistična fibroza se prenaša avtosomno recesivno. Gen za cistično fibrozo se nahaja v avtosomu 7, vsebuje 27 eksonov in je sestavljen iz 250.000 nukleotidnih parov.
En sam gen ima lahko veliko mutacij, od katerih je vsaka specifična za določeno populacijo ali geografsko regijo. Opisanih je bilo več kot 520 mutacij, najpogostejša med njimi je delta-P-508, tj. zamenjava aminokisline fenilalanina na položaju 508.
Patogeneza
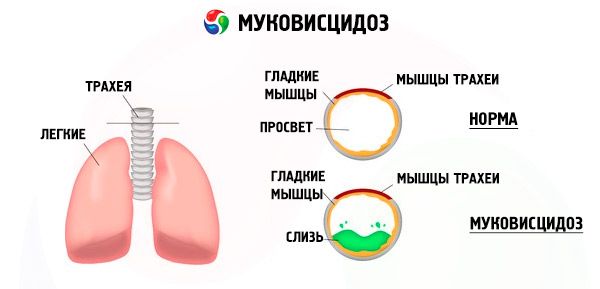
Mutacije v genu za cistično fibrozo motijo strukturo in delovanje beljakovine, imenovane CFTR (transmembranski regulator cistične fibroze). Ta beljakovina deluje kot kloridni kanal, ki sodeluje pri izmenjavi vode in elektrolitov v epitelijskih celicah bronhopulmonalnega sistema, prebavil, trebušne slinavke, jeter in reproduktivnega sistema. Zaradi motenega delovanja in strukture beljakovine CFTR se v celici kopičijo kloridni ioni Cl⁻ . To vodi do spremembe električnega potenciala v lumnu izločalnih kanalov, kar olajša pretok velikih količin natrijevih ionov (Na⁺ ) iz lumna kanala v celico in dodatno poveča absorpcijo vode iz pericelularnega prostora.
Zaradi teh sprememb se izločanje večine eksokrinih žlez zgosti, njegova evakuacija je motena, kar vodi do izrazitih sekundarnih motenj v organih in sistemih, najbolj izrazitih v bronhopulmonalnem in prebavnem sistemu.
V bronhih se razvije kronični vnetni proces različne intenzivnosti, delovanje ciliranega epitelija je močno moteno, sputum postane zelo viskozen, gost, zelo težko ga je izločiti, opazimo njegovo stagnacijo, nastanejo bronhiolo- in bronhiektazije, ki sčasoma postajajo vse pogostejše. Te spremembe vodijo do povečanja hipoksije in nastanka kronične pljučne srčne bolezni.
Bolniki s cistično fibrozo so izjemno nagnjeni k razvoju kroničnega vnetja v bronhopulmonalnem sistemu. To je posledica izrazitih motenj v lokalnem bronhopulmonalnem obrambnem sistemu (zmanjšane ravni IgA, interferona, fagocitne funkcije alveolarnih makrofagov in levkocitov).
Alveolarni makrofagi igrajo pomembno vlogo pri razvoju kroničnega vnetja v bronhopulmonalnem sistemu. Proizvajajo velike količine IL-8, kar dramatično poveča kemotaksijo nevtrofilcev v bronhialnem drevesu. Nevtrofilci se kopičijo v velikih količinah v bronhih in skupaj z epitelijskimi celicami izločajo številne provnetne citokine, vključno z IL-1, 8, 6, faktorjem tumorske nekroze in levkotrieni.
Pomembno vlogo pri patogenezi okvare bronhopulmonalnega sistema ima tudi visoka aktivnost encima elastaze. Ločimo med eksogeno in endogeno elastazo. Prvo proizvaja bakterijska flora (zlasti Pseudomonas aeruginosa), drugo pa nevtrofilni levkociti. Elastaza uničuje epitelij in druge strukturne elemente bronhijev, kar prispeva k nadaljnji motnji mukociliarnega transporta in hitremu nastanku bronhiektazij.
Nevtrofilni levkociti izločajo tudi druge proteolitične encime. Alfa-1-antipirzin in sekretorni zaviralec levkoproteinaz preprečujeta vpliv proteolitičnih encimov in s tem ščitita bronhopulmonalni sistem pred njihovim škodljivim vplivom. Vendar pa pri bolnikih s cistično fibrozo te zaščitne dejavnike žal zavira znatna količina nevtrofilne proteaze.
Vse te okoliščine prispevajo k vnosu okužbe v bronhopulmonalni sistem in razvoju kroničnega gnojnega bronhitisa. Poleg tega je treba upoštevati, da okvarjena beljakovina, ki jo kodira gen za cistično fibrozo, spremeni funkcionalno stanje bronhialnega epitelija, kar spodbuja adhezijo bakterij na bronhialni epitelij, predvsem Pseudomonas aeruginosa.
Poleg patologije bronhopulmonalnega sistema cistična fibroza povzroča tudi hude poškodbe trebušne slinavke, želodca, debelega in tankega črevesa ter jeter.
Simptomi cistična fibroza
Cistična fibroza se kaže z različnimi kliničnimi simptomi. Pri novorojenčkih se bolezen lahko kaže z mekonijevim ileusom. Zaradi pomanjkanja ali celo popolne odsotnosti tripsina mekonij postane zelo gost, viskozen in se kopiči v ileocekalnem predelu. Nadalje se razvije črevesna obstrukcija, ki se kaže z intenzivnim bruhanjem z dodatkom žolča, napihnjenostjo trebuha, pomanjkanjem izločanja mekonija, razvojem simptomov peritonitisa in hitrim povečanjem kliničnih manifestacij sindroma hude zastrupitve. Otrok lahko umre v prvih dneh življenja, če se ne izvede nujni kirurški poseg.
V manj hudih primerih je značilen znak cistične fibroze obilno, pogosto blato, mastno, z veliko količino maščobe, z zelo neprijetnim vonjem. Pri 1/3 bolnikov opazimo prolaps danke.
Posledično se pri bolnikih še naprej pojavljajo črevesna disfunkcija, sindrom malabsorpcije, hude motnje telesnega razvoja in huda hipovitaminoza.
V prvem ali drugem letu življenja se pojavijo simptomi poškodbe bronhopulmonalnega sistema (blaga oblika bolezni), ki se kaže v kašlju, ki je lahko izjemno izrazit in spominja na kašelj pri oslovskem kašlju. Kašelj spremljajo cianoza, zasoplost in izločanje gostega sputuma, sprva sluzastega, nato pa gnojnega. Postopoma se oblikuje klinična slika kroničnega obstruktivnega bronhitisa in bronhiektazij, pljučnega emfizema in respiratorne odpovedi. Otroci so izjemno dovzetni za akutne respiratorne virusne in bakterijske okužbe, kar prispeva k poslabšanju in napredovanju bronhopulmonalne patologije. Možen je razvoj bronhialne astme, odvisne od okužbe.
Pri šoloobveznih otrocih se cistična fibroza lahko kaže kot "črevesna kolika". Bolniki se pritožujejo nad hudimi paroksizmalnimi bolečinami v trebuhu, napihnjenostjo in ponavljajočim se bruhanjem. Pri palpaciji trebuha se določijo goste tvorbe, ki se nahajajo v projekciji debelega črevesa - blato, pomešano z gosto, gosto sluzjo. Otroci so zaradi prekomernega izločanja soli z znojem v vročem vremenu zelo nagnjeni k razvoju hipokloremične alkaloze, medtem ko se na otrokovi koži pojavi "slana zmrzal".
Bolezni bronhopulmonalnega sistema pri odraslih
Za okvaro bronhopulmonalnega sistema pri bolnikih s cistično fibrozo (pljučna oblika bolezni) je značilen razvoj kroničnega gnojnega obstruktivnega bronhitisa, bronhiektazij, kronične pljučnice, pljučnega emfizema, respiratorne odpovedi in pljučne srčne bolezni. Nekateri bolniki razvijejo pnevmotoraks in druge zaplete cistične fibroze: atelektazo, pljučne abscese, hemoptizo, pljučno krvavitev in od okužbe odvisno bronhialno astmo.
Bolniki se pritožujejo nad bolečim paroksizmalnim kašljem z zelo viskoznim, težko ločljivim sluzasto-gnojnim sputumom, včasih z dodatkom krvi. Poleg tega je izjemno značilna kratka sapa, najprej med telesno aktivnostjo, nato pa v mirovanju. Kratko sapo povzroča bronhialna obstrukcija. Mnogi bolniki se pritožujejo nad kroničnim rinitisom, ki ga povzročata polipoza in sinusitis. Značilna je tudi znatna šibkost, progresivno zmanjšanje zmogljivosti, pogoste akutne respiratorne virusne bolezni. Pri pregledu opazimo bledico kože, otekanje obraza, cianozo vidnih sluznic in hudo kratko sapo. Z razvojem dekompenzirane pljučne bolezni srca se v nogah pojavijo edemi. Opazimo lahko odebelitev končnih falang prstov v obliki bobnastih palčk in nohtov v obliki urnih stekelc. Prsni koš dobi obliko soda (zaradi razvoja pljučnega emfizema).
Tolkala pljuč razkrivajo znake emfizema - škatlast zvok, ostro omejitev gibljivosti pljučnega roba in znižanje spodnjega roba pljuč. Avskultacija pljuč razkriva ostro dihanje s podaljšanim izdihom, razpršeno suho piskanje ter vlažno srednje in drobno mehurčkasto piskanje. Pri hudem pljučnem emfizemu je dihanje močno oslabljeno.
Zunajpljučne manifestacije cistične fibroze
Zunajpljučne manifestacije cistične fibroze so lahko precej izrazite in se pojavljajo pogosto.
Poškodba trebušne slinavke
Pri 85 % bolnikov s cistično fibrozo opazimo različno hudo insuficienco eksokrine funkcije trebušne slinavke. Pri manjših poškodbah trebušne slinavke so sindromi maldigescije in malabsorpcije odsotni, prisotne so le laboratorijske manifestacije eksokrine insuficience (nizke ravni tripsina in lipaze v krvi in vsebini dvanajstnika; pogosto huda steatoreja). Znano je, da je za preprečevanje sindroma maldigescije zadostno izločanje le 1 do 2 % celotne lipaze. Klinično se kažejo le pomembne motnje eksokrine funkcije.
V normalnih pogojih acinus trebušne slinavke proizvajajo tekoči izloček, bogat z encimi. Ko se izloček premika po izločalnem kanalu, se obogati z vodo in anioni ter postane še bolj tekoč. Pri cistični fibrozi zaradi motnje v strukturi in delovanju transmembranskega regulatorja (kloridnega kanala) izloček trebušne slinavke ne prejme zadostne količine tekočine, postane viskozen, hitrost njegovega gibanja po izločalnem kanalu pa se močno upočasni. Beljakovine izločka se odlagajo na stene majhnih izločalnih kanalov, kar povzroči njihovo zaporo. Z napredovanjem bolezni se na koncu razvije uničenje in atrofija acinusov - nastane kronični pankreatitis z eksokrino insuficienco trebušne slinavke. To se klinično odraža v razvoju sindromov maldigescije in malabsorpcije. Insuficienca trebušne slinavke je glavni vzrok za malabsorpcijo maščob pri cistični fibrozi, vendar se to običajno opazi pri znatnem pomanjkanju lipaze. Forsher in Durie (1991) navajata, da se ob popolni odsotnosti pankreasne lipaze maščoba razgradi in absorbira za 50–60 %, kar je posledica prisotnosti želodčnih in sliničnih (sublingualnih) lipaz, katerih aktivnost je blizu spodnje meje norme. Ob motnjah v razgradnji in absorpciji maščob pride tudi do motenj v razgradnji in reabsorpciji beljakovin. Približno 50 % beljakovin, zaužitih s hrano, se izgubi z blatom. Absorpcija ogljikovih hidratov je kljub pomanjkanju α-amilaze prizadeta v manjši meri, vendar je lahko presnova ogljikovih hidratov znatno motena.
Poškodba trebušne slinavke se izraža v razvoju sindroma slabe prebave in malabsorpcije z znatno izgubo teže in obilnim mastnim blatom.
Razvoj sindromov maldigescije in malabsorpcije olajšajo tudi huda disfunkcija črevesnih žlez, moteno izločanje črevesnega soka in zmanjšanje vsebnosti črevesnih encimov v njem.
Sindromi maldigescije in malabsorpcije se imenujejo tudi črevesna oblika cistične fibroze.
Pri bolnikih s cistično fibrozo v poznih fazah bolezni (pri 2 % otrok in 15 % odraslih) opazimo okvarjeno endokrino delovanje trebušne slinavke (sladkorno bolezen).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Poškodbe jeter in žolčnih poti
Pri 13 % bolnikov z mešanimi in črevesnimi oblikami cistične fibroze se razvije ciroza jeter. Najbolj značilna je za mutacije W128X, delta-P508 in X1303K. Biliarna ciroza jeter s portalno hipertenzijo se odkrije pri 5–10 % bolnikov. Po Welchu, Smithu (1995) so klinični, morfološki, laboratorijski in instrumentalni znaki okvare jeter odkriti pri 86 % bolnikov s cistično fibrozo.
Mnogi bolniki s cistično fibrozo razvijejo tudi kronični holecistitis, pogosto kalkulozni.
Disfunkcija spolnih žlez
Bolniki s cistično fibrozo lahko doživijo azoospermijo, ki je vzrok za neplodnost. Zmanjšana plodnost je značilna tudi za ženske.
Faze
Obstajajo tri stopnje resnosti pljučne cistične fibroze.
Za blago obliko cistične fibroze so značilna redka poslabšanja (ne več kot enkrat na leto); v obdobjih remisije so klinične manifestacije praktično odsotne in bolniki so sposobni za delo.
Zmerna resnost - poslabšanja se pojavijo 2-3 krat na leto in trajajo približno 2 meseca ali dlje. V fazi poslabšanja se pojavijo intenziven kašelj s težko izločljivim sputumom, zasoplost tudi pri manjših fizičnih naporih, subfebrilna telesna temperatura, splošna šibkost, potenje. Hkrati pride do kršitve eksokrine funkcije trebušne slinavke. V fazi remisije se delovna zmogljivost ne obnovi v celoti, zasoplost med fizičnim naporom ostaja.
Za hud potek so značilna zelo pogosta poslabšanja bolezni. Remisije praktično niso prisotne. V klinični sliki pridejo v ospredje huda respiratorna odpoved, simptomi kronične pljučne srčne bolezni, pogosto dekompenzirane, značilna je hemoptiza. Opažena je znatna izguba teže, bolniki so popolnoma invalidni. Praviloma hudo bronhopulmonalno patologijo spremlja močno izražena disfunkcija trebušne slinavke.
Obrazci
- Bronhopulmonalne lezije
- Ponavljajoča se in ponavljajoča se pljučnica z dolgotrajnim potekom.
- Abscesirajoča pljučnica, zlasti pri dojenčkih.
- Kronična pljučnica, zlasti dvostranska.
- Bronhialna astma, odporna na tradicionalno zdravljenje.
- Ponavljajoči se bronhitis, bronhiolitis, zlasti s kulturo Pseudomonas aeruginosa.
- Spremembe v prebavilih
- Mekonijev ileus in njegovi ekvivalenti.
- Sindrom motene črevesne absorpcije neznanega izvora.
- Obstruktivna zlatenica pri novorojenčkih s podaljšanim potekom.
- Ciroza jeter.
- Sladkorna bolezen.
- Gastroezofagealni refluks.
- Holelitiaza.
- Rektalni prolaps.
- Spremembe v drugih organih in sistemih
- Motnje rasti in razvoja.
- Zapoznel spolni razvoj.
- Moška neplodnost.
- Nosni polipi.
- Bratje in sestre iz družin s cistično fibrozo.
[ 24 ]
Zapleti in posledice
Zapleti iz prebavil vključujejo:
- Sladkorna bolezen se razvije pri 8–12 % bolnikov, starejših od 25 let.
- Fibrotična kolonopatija.
- Mekonijev ileus v neonatalnem obdobju (pri 12 % novorojenčkov s cistično fibrozo), sindrom distalne črevesne obstrukcije, rektalni prolaps, peptični ulkus in gastroezofagealna refluksna bolezen.
Zapleti z jetri:
- Zamaščenost jeter (pri 30–60 % bolnikov),
- Fokalna biliarna ciroza, multinodularna biliarna ciroza in z njo povezana portalna hipertenzija.
Portalna hipertenzija včasih povzroči smrt zaradi varic požiralnika.
Razširjenost holecistitisa in žolčnih kamnov je pri bolnikih s cistično fibrozo večja kot pri drugih posameznikih.
Zapoznela puberteta in zmanjšana plodnost ter drugi zapleti. Večina moških ima azoospermijo in nerazvitost semenovoda.
Diagnostika cistična fibroza
Splošna krvna analiza - tipična je anemija različne stopnje, običajno normo- ali hipokromna. Anemija ima polifaktorsko genezo (zmanjšana absorpcija železa in vitamina B12 v črevesju zaradi razvoja malabsorpcijskega sindroma). Možna je levkopenija, z razvojem gnojnega bronhitisa in pljučnice - levkocitoza, povečana sedimentacija eritrocitov.
Splošna analiza urina - brez pomembnih sprememb, v redkih primerih opazimo rahlo proteinurijo.
Koprološki pregled - opazimo steatorejo, kreatorejo. Becker (1987) priporoča merjenje himotripsina in maščobnih kislin v blatu. Pred določanjem himotripsina v blatu je treba vsaj 3 dni pred preiskavo prenehati jemati prebavne encime. Pri cistični fibrozi je količina himotripsina v blatu zmanjšana, količina maščobnih kislin pa povečana (normalno izločanje maščobnih kislin je manjše od 20 mmol/dan). Upoštevati je treba, da se povečano izločanje maščobnih kislin z blatom opazi tudi pri:
- pomanjkanje konjugiranih maščobnih kislin v tankem črevesu zaradi odpovedi jeter, obstrukcije žolčnih vodov, znatne bakterijske kolonizacije tankega črevesa (v tem primeru pride do intenzivne hidrolize žolčnih kislin);
- ileitis;
- celiakija (z razvojem malabsorpcijskega sindroma);
- enteritis;
- črevesni limfomi;
- Whippleova bolezen;
- alergije na hrano;
- pospešen tranzit mase hrane pri driski različnega izvora, karcinoidnem sindromu, tirotoksikozi.
Biokemični krvni test - znižana raven skupnih beljakovin in albuminov, zvišana raven alfa2 in gama globulinov, bilirubina in aminotransferaz (v primeru okvare jeter), zmanjšana aktivnost amilaze, lipaze, tripsina ter ravni železa in kalcija (v primeru razvoja sindroma maldigescije, malabsorpcije).
Analiza sputuma - prisotnost velikega števila nevtrofilnih levkocitov in mikroorganizmov (med bakterioskopijo sputuma).
Študija absorpcijske funkcije tankega črevesa in eksokrine funkcije trebušne slinavke razkriva pomembne motnje.
Rentgenski pregled pljuč - razkrije spremembe, katerih resnost je odvisna od resnosti in faze bolezni. Najbolj značilne spremembe so:
- povečano pljučno vzorčenje zaradi peribronhialnih intersticijskih sprememb;
- širitev korenin pljuč;
- slika lobularne, subsegmentalne ali celo segmentne atelektaze pljuč;
- povečana prosojnost pljučnih polj, predvsem v zgornjih delih, nizek položaj in nezadostna gibljivost diafragme, širitev retrosternalnega prostora (manifestacija pljučnega emfizema);
- segmentna ali polisegmentalna infiltracija pljučnega tkiva (pri razvoju pljučnice).
Bronhografija razkriva spremembe, ki jih povzroča obstrukcija bronhijev z viskoznim sputumom (razdrobljenost polnjenja bronhijev s kontrastom, neenakomerne konture, pojav rupture bronhijev, znatno zmanjšanje števila stranskih vej), pa tudi bronhoekgamije (valjaste, mešane), lokalizirane predvsem v spodnjih delih pljuč.
Bronhoskopija razkriva difuzni gnojni bronhitis z obilnim gostim, viskoznim sputumom in fibrinoznimi filmi.
Spirometrija - že v zgodnjih fazah bolezni razkrije dihalno odpoved obstruktivnega tipa (zmanjšan FVC, FEV1, Tiffnov indeks), restriktivno (zmanjšan FVC) ali najpogosteje obstruktivno-restriktivno (zmanjšan FVC, FVC, FEV1, Tiffnov indeks).
Gibsonov in Cookov test znoja (test elektrolitov v znoju) vključuje spodbujanje znojenja z elektroforezo pilokarpina, ki ji sledi določanje kloridov v znoju. Doerehuk (1987) opisuje test na naslednji način. Elektroforeza pilokarpina se izvaja na podlakti, električni tok je 3 mA. Po čiščenju kože z destilirano vodo se znoj zbere s filtrirnim papirjem, ki se namesti na stimulirano območje in prekrije z gazo, da se prepreči izhlapevanje. Po 30–60 minutah se filtrirni papir odstrani in eluira z destilirano vodo. Izmeri se količina zbranega znoja. Za zanesljive rezultate je treba zbrati vsaj 50 mg (po možnosti 100 mg) znoja.
Če je koncentracija klorida večja od 60 mmol/l, se diagnoza cistične fibroze šteje za verjetno; če je koncentracija klorida večja od 100 mmol/l, je zanesljiva; v tem primeru razlika v koncentraciji klora in natrija ne sme presegati 8-10 mmol/l. Hadson (1983) priporoča, da se v primeru mejne vsebnosti natrija in klorida v znoju opravi prednizolonski test (5 mg peroralno 2 dni, nato pa določitev elektrolitov v znoju). Pri posameznikih, ki ne trpijo za cistično fibrozo, se raven natrija v znoju zniža na spodnjo mejo normale; pri cistični fibrozi se ne spremeni. Znojni test je priporočljiv za vsakega otroka s kroničnim kašljem.
Analiza krvnih madežev ali vzorcev DNK za odkrivanje večjih mutacij gena za cistično fibrozo je najbolj občutljiv in specifičen diagnostični test. Vendar je ta metoda primerna le za države, kjer je stopnja mutacije delta-P508 višja od 80 %. Poleg tega je tehnika zelo draga in tehnično zapletena.
Prenatalna diagnoza cistične fibroze se izvaja z določanjem izoencimov alkalne fosfataze v amnijski tekočini. Ta metoda postane mogoča od 18. do 20. tedna nosečnosti.
Glavna merila za diagnozo cistične fibroze so naslednja:
- indikacije v anamnezi zaostalega telesnega razvoja v otroštvu, ponavljajoče se kronične bolezni dihal, dispeptične motnje in driska, prisotnost cistične fibroze pri bližnjih sorodnikih;
- kronični obstruktivni bronhitis, pogosto ponavljajoč se, z razvojem bronhiektazij in pljučnega emfizema, pogosto ponavljajoča se pljučnica;
- kronični ponavljajoči se pankreatitis z izrazitim zmanjšanjem eksokrine funkcije, sindrom malabsorpcije;
- povečana vsebnost klora v bolnikovem znoju;
- neplodnost z ohranjeno spolno funkcijo.
Uspešno diagnozo in diferencialno diagnozo cistične fibroze olajša identifikacija rizičnih skupin.
Program presejanja za cistično fibrozo
- Splošna analiza krvi, urina, sputuma.
- Bakteriološka analiza sputuma.
- Koprološka analiza.
- Biokemijski krvni test: določanje skupnih beljakovin in beljakovinskih frakcij, glukoze, bilirubina, aminotransferaz, alkalne fosfataze, gama-glutamil transpeptidaz, kalija, kalcija, železa, lipaze, amilaze, tripsina.
- Študija eksokrine funkcije trebušne slinavke in absorpcijske funkcije črevesja.
- Fluoroskopija in radiografija pljuč, CT pljuč.
- EKG.
- Ehokardiografija.
- Bronhoskopija in bronhografija.
- Spirometrija.
- Test znojenja.
- Posvetovanje z genetikom.
- Analiza krvnih madežev ali vzorcev DNK za večje mutacije gena za cistično fibrozo.
Kaj je treba preveriti?
Katere teste so potrebne?
Koga se lahko obrnete?
Zdravljenje cistična fibroza
Vrsta in resnost simptomov cistične fibroze se lahko zelo razlikujeta, zato ni tipičnega načrta zdravljenja; zdravljenje je individualizirano za vsakega posameznika.
Terapija je sestavljena iz naslednjih terapevtskih ukrepov:
- Dihalne vaje in posturalna drenaža pomagajo odpraviti gosto sluz, ki se nabira v pljučih. Nekatere tehnike čiščenja dihalnih poti zahtevajo pomoč družinskih članov, prijateljev ali pulmologa. Mnogi ljudje uporabljajo napihljiv prsni jopič, ki vibrira z visoko frekvenco.

- Inhalacijska zdravila, ki imajo bronhodilatatorne, drenažne (mukolitiki) in antibakterijske učinke (na primer fluorokinoloni).
- Pripravki, ki vsebujejo trebušne slinavke in encime za izboljšanje prebave. Ti pripravki se jemljejo med obroki.
- Multivitamini (vključno z vitamini, topnimi v maščobah).
Leta 2015 je FDA odobrila drugo zdravilo za zdravljenje cistične fibroze, ki cilja na okvarjeno beljakovino, znano kot CFTR. Prvo zdravilo, tako imenovani modulator CFTR, je bilo odobreno leta 2012. Pričakuje se, da bodo modulatorji CFTR nekaterim ljudem s cistično fibrozo podaljšali življenje za desetletja.
Za zdravljenje naslednjih respiratornih zapletov je lahko potreben kirurški poseg:
- Pnevmotoraks, masivna ponavljajoča se ali vztrajna hemoptiza, nosni polipi, vztrajni in kronični sinusitis.
- Mekonijev ileus, invaginacija, rektalni prolaps.
Presaditev pljuč se izvaja v terminalni fazi bolezni.
Napoved
Povprečna starost preživetja bolnikov s cistično fibrozo se giblje od 35 do 40 let. Povprečna starost preživetja je pri moških višja kot pri ženskah.
S sodobnimi strategijami zdravljenja 80 % bolnikov doseže odraslost. Vendar pa cistična fibroza znatno omejuje bolnikove funkcionalne sposobnosti. Za to bolezen še vedno ni zdravila.