Medicinski strokovnjak članka

Nove publikacije
Charcot-Marie-Tooth bolezen
Zadnji pregled: 23.11.2021

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
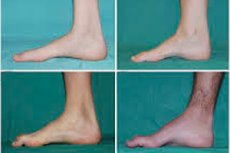
Atrofija, sindrom peronealne mišice ali Charcot-Marie-Toothova bolezen je cela skupina kroničnih dednih bolezni s poškodbami perifernih živcev.
V skladu z ICD-10 v oddelku bolezni živčnega sistema je koda te bolezni G60.0 (dedna motorična in senzorična nevropatija). Vključen je tudi na seznam sirot.
Epidemiologija
Po klinični statistiki je razširjenost vseh vrst Charcot-Marie-Tooth bolezni na 100 tisoč prebivalcev 19 primerov (po drugih virih en primer na 2,5-10 tisoč prebivalcev).
CMT tipa 1 predstavlja približno dve tretjini primerov (en primer na 5-7 tisoč prebivalcev) in skoraj 70% jih je povezanih s podvajanjem gena PMP22. Na svetu ta vrsta bolezni prizadene več kot 1,2 milijona ljudi.
Incidenca CMT tipa 4 je ocenjena na 1-5 primerov na 10 tisoč otrok. [1]
Vzroki charcot-Marie-Tooth bolezen
Po klasifikaciji polinevropatskih sindromov se atrofija peronealnih (peronealnih) mišic, živčna amiotrofija Charcot-Marie-Tooth ali Charcot-Marie-Toothova bolezen (okrajšana kot CMT) nanaša na gensko določene motorično senzorične polinevropatije. [2]
To pomeni, da so razlogi za njegov nastanek genetske mutacije. In glede na naravo genetskih nepravilnosti se glavne vrste ali tipi tega sindroma razlikujejo: demielinizirajoči in aksonski. V prvo skupino spada bolezen Charcot-Marie-Tooth tipa 1 (CMT1), ki se pojavi kot posledica podvajanja gena PMP22 na kromosomu 17, ki kodira transmembranski periferni mielinski protein 22. Posledično segmentarna demielinizacija aksonske ovojnice (procesi živčnih celic) in pride do zmanjšanja hitrosti prevodnosti živcev. Poleg tega lahko pride do mutacij nekaterih drugih genov.
Aksonska oblika je Charcot-Marie-Toothova bolezen tipa 2 (CMT2), ki prizadene same aksone in je povezana s patološkimi spremembami gena MFN2 na loku 1p36.22, ki kodira membranski protein mitofusin-2, kar je potrebno za fuzijo mitohondrijev in oblikovanje funkcionalnih mitohondrijskih mrež znotraj perifernih živcev celic. Obstaja več kot ducat podtipov CMT2 (z mutacijami v določenih genih).
Treba je opozoriti, da je zdaj identificiranih več kot sto genov, katerih dedna škoda povzroča različne podtipe Charcot-Marie-Tooth bolezni. Na primer, mutacije v genu RAB7 razvijejo CMT tipa 2B; sprememba gena SH3TC2 (ki kodira enega od proteinov Schwannovih membran celic) povzroči CMT tipa 4C, ki se kaže v otroštvu in je zanj značilna demielinizacija motoričnih in senzoričnih nevronov (pol ducata oblik tipa 4 to bolezen razlikujejo).
V zgodnjem otroštvu se začne razvijati tudi redek SMT tipa 3 (imenovan Dejerine-Sott sindrom), ki ga povzročajo mutacije genov PMP22, MPZ, EGR2 in drugih.
Ko se CMT tipa 5 pojavi v starosti 5-12 let, ni opažena le motorična nevropatija (v obliki spastične parapareze spodnjih okončin), temveč tudi poškodba optičnega in slušnega živca.
Za CMT tipa 6 so značilne mišična oslabelost in atrofija vidnega živca (z izgubo vida) ter težave z ravnotežjem. In pri tipu 7 Charcot-Marie-Toothove bolezni ne opazimo le motorično-senzorične nevropatije, temveč tudi bolezni mrežnice v obliki pigmentnega retinitisa.
Pogostejša X-vezana SMT ali Charcot-Marie-Toothova bolezen s tetraparezo okončin (oslabitev gibanja rok in nog) pri moških je demielinizirajočega tipa in velja za rezultat mutacije gena GJB1 na dolg krak X-kromosoma, ki kodira za Connexin 32, transmembranski protein Schwannove celice in oligodendrocite, ki uravnava prenos živčnih signalov. [3]
Dejavniki tveganja
Glavni dejavnik tveganja za CMT je prisotnost te bolezni v družinski anamnezi, torej pri bližnjih sorodnikih.
Po mnenju genetikov je, če sta oba starša nosilca avtosomno recesivnega gena Charcot-Marie-Tooth bolezni, tveganje za otroka, ki bo razvil to bolezen, 25%. Tveganje, da bo otrok nosil ta gen (sam pa ne bo imel nobenih simptomov), je ocenjeno na 50%.
V primeru dedovanja, povezanega s X (kadar je mutirani gen na ženskem X-kromosomu), obstaja 50-odstotno tveganje, da bo mati ta gen prenesla na svojega sina in bo razvil CMT bolezen. Ko se rodi ženski otrok, se bolezen morda ne pojavi, vendar hčerini sinovi (vnuki) lahko podedujejo okvarjeni gen - z razvojem bolezni.
Patogeneza
Pri kateri koli vrsti Charcot-Marie-Tooth bolezni je njena patogeneza posledica dedne anomalije perifernih živcev: motorja (motorja) in senzorja (senzorja).
Če je tip CMT demielinizirajoč, potem uničenje ali okvara mielinske ovojnice, ki ščiti aksone perifernih živcev, povzroči upočasnitev prenosa živčnih impulzov perifernega živčnega sistema - med možgane, mišice in senzorične organe.
Pri aksonskem tipu bolezni so aksoni neposredno prizadeti, kar negativno vpliva na moč živčnih signalov, kar je nezadostno za popolno stimulacijo mišic in senzoričnih organov.
Preberite tudi:
Kako se širi Charcot-Marie-Toothov sindrom? Okvareni geni se lahko podedujejo avtosomno dominantno ali avtosomno recesivno.
Najpogosteje - avtosomno dominantno dedovanje - se pojavi, če obstaja ena kopija mutiranega gena (ki ga nosi eden od staršev). Verjetnost prenosa CMT na vsakega od rojenih otrok je ocenjena na 50%. [4]
Pri avtosomno recesivnem dedovanju bolezen zahteva dve kopiji okvarjenega gena (po enega od vsakega starša, ki nima znakov bolezni).
V 40-50% primerov pride do avtosomno dominantne dedne demielinizacije, to je CMT tipa 1; v 12-26% primerov - aksonski CMT, to je tip 2. In v 10-15% primerov opazimo X-vezano dedovanje. [5]
Simptomi charcot-Marie-Tooth bolezen
Običajno se prvi znaki te bolezni začnejo pojavljati v otroštvu in mladostnikih ter se postopoma razvijajo skozi vse življenje, čeprav se sindrom lahko počuti pozneje. Kombinacija simptomov je spremenljiva in hitrosti napredovanja bolezni ter njene resnosti ni mogoče predvideti.
Obstajajo tipični simptomi začetne faze, kot je povečana splošna utrujenost; zmanjšan tonus (šibkost) mišic stopal, gležnjev in spodnjih nog; pomanjkanje refleksov. To otežuje premikanje stopala in vodi do disbazije (motnje hoje) v obliki višjega dviga nog, pogosto s pogostim spotikanjem in padanjem. Znaki bolezni Charcot-Marie-Tooth pri majhnem otroku so lahko izrazita okornost in težave pri hoji, nenavadne za starost, povezane z dvostranskim visečim stopalom . Značilne so tudi deformacije stopala: visok lok (votla noga) ali močna ploska stopala, ukrivljeni (kladivu podobni) prsti.
V primeru hoje na prstih v ozadju mišične hipotenzije lahko nevrolog sumi, da ima otrok CMT tip 4, pri katerem otroci do mladosti morda ne bodo mogli več hoditi.
Z napredovanjem se mišična atrofija in šibkost širijo na zgornje okončine, kar otežuje fine motorične sposobnosti in običajne aktivnosti rok. Zmanjšanje otipnih občutkov in sposobnost občutka toplote in mraza ter otrplost stopal in rok kaže na poškodbe aksonov čutnih živcev.
Z manifestacijo v otroštvu Charcot-Marie-Tooth bolezni 3 in 6 tipa obstaja občutljiva ataksija (motena koordinacija gibov in ravnotežja), mišično trzanje in tresenje, poškodba obraznega živca, atrofija optičnega očesa z nistagmusom, izguba sluha.
V kasnejših fazah lahko pride do neobvladljivega tresenja (tresenja) in pogostih mišičnih krčev; težave z gibanjem lahko privedejo do razvoja bolečine: mišične, sklepne, nevropatske.
Zapleti in posledice
Charcot-Marie-Toothova bolezen ima lahko zaplete in posledice, kot so:
- pogostejši zvini in zlomi;
- kontrakture, povezane s krajšanjem periartikularnih mišic in kit;
- skolioza (ukrivljenost hrbtenice);
- težave z dihanjem - s poškodbami živčnih vlaken, ki inervirajo mišice trebušne prepone:
- izguba sposobnosti samostojnega gibanja.
Diagnostika charcot-Marie-Tooth bolezen
Diagnostika vključuje klinični pregled, anamnezo (vključno z družinsko anamnezo), nevrološki in sistemski pregled.
Preskusi se izvajajo za preverjanje obsega gibanja, občutljivosti in tetivnih refleksov. Prevodnost živcev lahko ocenimo z instrumentalno diagnostiko - elektromiografijo ali elektronevromiografijo . Morda bo potreben tudi ultrazvok ali magnetna resonanca. [6]
Genetska ali DNK diagnostika za prepoznavanje najpogostejših genetskih mutacij, ki povzročajo CMT na vzorcu krvi, je omejena, saj DNK testi trenutno niso na voljo za vse vrste CMT. Za podrobnosti glej - Genetske raziskave
V nekaterih primerih se opravi biopsija perifernega živca (običajno gastrocnemiusa).
Diferencialna diagnoza
Diferencialna diagnoza se izvaja z drugimi perifernimi nevropatijami, mišično distrofijo Duchenne, mielopatskim in miasteničnim sindromom, diabetično nevropatijo, mieloaptijami v primeru multiple in amiotrofične lateralne skleroze, Guillain-Barréjevega sindroma, travme peronealnega živca in atrofij diska hrbtenice ), poškodbe malih možganov ali talamusa ter neželeni učinki kemoterapije (kadar se zdravijo s citostatiki, kot sta vinkristin ali paklitaksel). [7]
Koga se lahko obrnete?
Zdravljenje charcot-Marie-Tooth bolezen
Danes je zdravljenje te dedne bolezni sestavljeno iz fizioterapevtskih vaj (namenjenih krepitvi in raztezanju mišic); delovna terapija (ki pomaga bolnikom z mišično oslabelostjo v rokah); z uporabo ortopedskih pripomočkov za lažjo hojo. Če je potrebno, vzemite zdravila proti bolečinam ali antikonvulzive. [8]
V primeru izrazite ploskosti lahko izvedemo osteotomijo, v primeru deformacije pete pa je indicirana njihova kirurška korekcija - artrodeza. [9]
Potekajo raziskave tako o genski komponenti bolezni kot o načinih njenega zdravljenja. Uporaba izvornih celic, nekaterih hormonov, lecitina ali askorbinske kisline še ni dala pozitivnih rezultatov.
Toda zahvaljujoč najnovejšim raziskavam se lahko v bližnji prihodnosti resnično pojavi nova pri zdravljenju Charcot-Marie-Tooth bolezni. Tako od leta 2014 francosko podjetje Pharnext razvija, od sredine leta 2019 pa klinična preskušanja zdravila PXT3003 za zdravljenje CMT tipa 1 pri odraslih, ki zavira povečano izražanje gena PMP22, izboljšuje mieliniranje perifernih živcev in oslabitev živčno-mišičnih simptomov.
Strokovnjaki medicinske družbe Sarepta Therapeutics (ZDA) delajo na genski terapiji za bolezen Charcot-Marie-Tooth tipa 1. Pri tej terapiji bomo uporabili neškodljiv z adenom povezan virus (AAV) iz rodu Dependovirus z linearnim enoverižnim genomom DNA, ki bo v telo prenesel gen NTF3, ki kodira protein nevrotropin-3 (NT-3), potreben za delovanje Schwannovih živčnih celic.
Do konca leta 2020 bo Helixmith začel klinična preskušanja genske terapije Engensis (VM202), razvite v Južni Koreji za zdravljenje mišičnih simptomov pri CMT tipa 1. [10]
Preprečevanje
Preprečevanje CMT je lahko genetsko svetovanje bodočim staršem, še posebej, če ima kdo iz zakonskega para to bolezen v družini. Ugotovljeni pa so bili primeri mutacij genskih točk de novo, torej v odsotnosti bolezni v družinski anamnezi.
Med nosečnostjo vzorčenje horionskih resic (od 10 do 13 tednov nosečnosti) in analiza plodovnice (v 15-18 tednih) omogoča preverjanje verjetnosti Charcot-Marie-Tooth bolezni pri nerojenem otroku.
Napoved
Na splošno so napovedi za različne vrste bolezni Charcot-Marie-Tooth odvisne od klinične resnosti, v vsakem primeru pa bolezen napreduje počasi. Mnogi bolniki imajo invalidnost, čeprav to ne zmanjša pričakovane življenjske dobe.