Medicinski strokovnjak članka

Nove publikacije
Bolezen Charcot-Marie-Tooth.
Zadnji pregled: 12.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
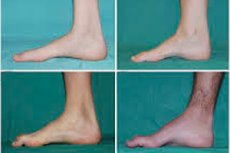
Peronealna mišična atrofija, Charcot-Marie-Toothov sindrom ali bolezen, je skupina kroničnih dednih bolezni s poškodbo perifernih živcev.
Po MKB-10 je v poglavju o boleznih živčnega sistema koda za to bolezen G60.0 (dedna motorična in senzorična nevropatija). Uvrščena je tudi na seznam bolezni sirot.
Epidemiologija
Glede na klinično statistiko je razširjenost vseh vrst bolezni Charcot-Marie-Tooth na 100 tisoč prebivalcev 19 primerov (po drugih virih en primer na 2,5-10 tisoč prebivalcev).
CMT tip 1 predstavlja približno dve tretjini primerov (en primer na 5000 do 7000 prebivalcev), skoraj 70 % jih je povezanih s podvajanjem gena PMP22. Za to vrsto bolezni trpi več kot 1,2 milijona ljudi po vsem svetu.
Incidenca karcinogenne možganske moteče bolezni tipa 4 je ocenjena na 1–5 primerov na 10.000 otrok. [ 1 ]
Vzroki Bolezen Charcot-Marie-Tooth
Glede na klasifikacijo polinevropatskih sindromov se peronealna (fibularna) mišična atrofija, Charcot-Marie-Toothova nevronska amiotrofija ali Charcot-Marie-Toothova bolezen (okrajšano CMT) nanaša na genetsko določene motorično-senzorične polinevropatije. [ 2 ]
To pomeni, da so vzroki za njegov pojav genetske mutacije. Glede na naravo genetskih odstopanj ločimo glavne vrste ali vrste tega sindroma: demielinizirajočo in aksonsko. Prva skupina vključuje Charcot-Marie-Toothovo bolezen tipa 1 (CMT1), ki nastane zaradi podvajanja gena PMP22 na kromosomu 17, ki kodira transmembranski periferni mielinski protein 22. Posledično pride do segmentne demielinizacije aksonske ovojnice (odrastkov živčnih celic) in zmanjšanja hitrosti prevajanja živčnih signalov. Poleg tega so lahko mutacije prisotne tudi v nekaterih drugih genih.
Aksonska oblika je Charcot-Marie-Toothova bolezen tipa 2 (CMT2), ki prizadene same aksone in je povezana s patološkimi spremembami gena MFN2 na lokusu 1p36.22, ki kodira membranski protein mitofuzin-2, ki je potreben za fuzijo mitohondrijev in nastanek funkcionalnih mitohondrijskih mrež znotraj perifernih živčnih celic. Obstaja več kot ducat podtipov CMT2 (z mutacijami v specifičnih genih).
Treba je opozoriti, da je bilo trenutno identificiranih več kot sto genov, katerih poškodbe, ki se prenašajo z dedovanjem, povzročajo različne podtipe Charcot-Marie-Toothove bolezni. Na primer, mutacije v genu RAB7 vodijo do CMT tipa 2B; sprememba gena SH3TC2 (ki kodira enega od membranskih proteinov Schwannovih celic) povzroči CMT tipa 4C, ki se kaže v otroštvu in je značilna po demielinizaciji motoričnih in senzoričnih nevronov (obstaja ducat in pol oblik te bolezni tipa 4).
Redka karpometakarpulmonalna motnja tipa 3 (imenovana Dejerine-Sottasov sindrom) se začne razvijati v zgodnjem otroštvu in jo povzročajo mutacije v genih PMP22, MPZ, EGR2 in drugih.
Ko se CMT tipa 5 pojavi v starosti 5-12 let, se ne opazi le motorična nevropatija (v obliki spastične parapareze spodnjih okončin), temveč tudi poškodba vidnega in slušnega živca.
Mišična šibkost in optična atrofija (z izgubo vida) ter težave z ravnotežjem so značilne za CMT tipa 6. Pri Charcot-Marie-Toothovi bolezni tipa 7 ni prisotna le motorično-senzorična nevropatija, temveč tudi bolezen mrežnice v obliki retinitis pigmentosa.
Pogostejša pri moških je na X vezana CMT ali Charcot-Marie-Toothova bolezen s tetraparezo okončin (šibkost gibanja rok in nog), ki je demielinizirajočega tipa in naj bi nastala zaradi mutacije v genu GJB1 na dolgem kraku kromosoma X, ki kodira koneksin 32, transmembranski protein Schwannovih celic in oligodendrocitov, ki uravnava prenos živčnih signalov. [ 3 ]
Dejavniki tveganja
Glavni dejavnik tveganja za CMT je družinska anamneza bolezni, torej pri bližnjih sorodnikih.
Po mnenju genetikov je tveganje za otroka, ki bo razvil to bolezen, 25 %, če sta oba starša nosilca avtosomno recesivnega gena za Charcot-Marie-Toothovo bolezen, tveganje, da bo otrok nosilec tega gena (vendar ne bo imel nobenih simptomov), pa je ocenjeno na 50 %.
V primeru dedovanja, vezanega na X (ko se mutirani gen nahaja na kromosomu X ženske), obstaja 50-odstotno tveganje, da bo mati gen prenesla na sina, ki bo razvil karpometakarpulmonalno melanocitno bolezen (CMT). Bolezen se morda ne bo pojavila ob rojstvu deklice, vendar lahko hčerini sinovi (vnuki) podedujejo okvarjeni gen in bolezen se bo razvila.
Patogeneza
Pri kateri koli vrsti Charcot-Marie-Toothove bolezni je njena patogeneza posledica dedne anomalije perifernih živcev: motoričnih (gibalnih) in senzoričnih (občutljivih).
Če je tip CMT demielinizirajoč, potem uničenje ali okvara mielinske ovojnice, ki ščiti aksone perifernih živcev, povzroči upočasnitev prenosa živčnih impulzov v perifernem živčnem sistemu - med možgani, mišicami in čutilnimi organi.
Pri aksonski vrsti bolezni so prizadeti sami aksoni, kar negativno vpliva na moč živčnih signalov, ki je nezadostna za popolno stimulacijo mišic in čutnih organov.
Preberite tudi:
Kako se prenaša Charcot-Marie-Toothov sindrom? Okvarjeni geni se lahko dedujejo avtosomno dominantno ali avtosomno recesivno.
Najpogostejši tip, avtosomno dominantno dedovanje, se pojavi, ko obstaja ena kopija mutiranega gena (ki ga nosi eden od staršev). Verjetnost prenosa CMT na vsakega rojenega otroka je ocenjena na 50 %. [ 4 ]
Pri avtosomno recesivnem dedovanju sta za razvoj bolezni potrebni dve kopiji okvarjenega gena (po ena od vsakega starša, ki ne kaže nobenih znakov bolezni).
V 40–50 % primerov se pojavi avtosomno dominantna dedna demielinizacija, tj. karpometakarpulmonalna možganska mielinizacija tipa 1; v 12–26 % primerov aksonska karpometakarpulmonalna možganska mielinizacija, tj. tip 2. V 10–15 % primerov pa opazimo dedovanje, vezano na X. [ 5 ]
Simptomi Bolezen Charcot-Marie-Tooth
Običajno se prvi znaki te bolezni začnejo pojavljati v otroštvu in adolescenci ter se postopoma razvijajo skozi vse življenje, čeprav se sindrom lahko pojavi tudi kasneje. Kombinacija simptomov je spremenljiva, hitrost napredovanja bolezni in njeno resnost pa ni mogoče napovedati.
Tipični simptomi začetne faze vključujejo povečano splošno utrujenost; zmanjšan tonus (šibkost) mišic stopal, gležnjev in goleni; pomanjkanje refleksov. To otežuje gibanje stopal in vodi do disbazije (motnje hoje) v obliki višjega dviga nog, pogosto s pogostimi spotikanji in padci. Znaki Charcot-Marie-Toothove bolezni pri majhnem otroku lahko vključujejo izrazito nerodnost in starostno neznačilne težave s hojo, povezane z obojestranskim spuščanjem stopala. Značilne so tudi deformacije stopal: visok lok (votlo stopalo) ali huda ploska stopala, ukrivljeni (kladivasti) prsti.
V primeru hoje po prstih na ozadju mišične hipotonije lahko nevrolog posumi, da ima otrok CMT tipa 4, pri kateri otroci do adolescence morda ne bodo mogli več hoditi.
Z napredovanjem bolezni se atrofija in šibkost mišic širita na zgornje okončine, zaradi česar je težko izvajati finomotorične sposobnosti in običajna opravila z rokami. Zmanjšani taktilni občutki in sposobnost občutenja toplote in mraza ter otrplost v stopalih in rokah kažejo na poškodbo aksonov senzoričnih živcev.
Pri Charcot-Marie-Toothovi bolezni tipa 3 in 6, ki se kaže v otroštvu, opazimo senzorično ataksijo (moteno koordinacijo gibov in ravnotežja), trzanje mišic in tremor, poškodbo obraznega živca, atrofijo vidnega živca z nistagmusom in izgubo sluha.
V kasnejših fazah se lahko pojavijo neobvladljivo tresenje (tremor) in pogosti mišični krči; težave z gibanjem lahko vodijo do razvoja bolečin: mišičnih, sklepnih, nevropatskih.
Zapleti in posledice
Charcot-Marie-Toothova bolezen ima lahko zaplete in posledice, kot so:
- pogostejši zvini in zlomi;
- kontrakture, povezane s skrajšanjem periartikularnih mišic in tetiv;
- skolioza (ukrivljenost hrbtenice);
- težave z dihanjem – ko so poškodovana živčna vlakna, ki oživčujejo mišice diafragme:
- izguba sposobnosti samostojnega gibanja.
Diagnostika Bolezen Charcot-Marie-Tooth
Diagnoza vključuje klinični pregled, anamnezo (vključno z družinsko anamnezo), nevrološki in sistemski pregled.
Izvajajo se testi za preverjanje obsega gibanja, občutljivosti in tetivnih refleksov. Prevodnost živcev je mogoče oceniti z instrumentalno diagnostiko – elektromiografijo ali elektronevromiografijo. Morda bo potreben tudi ultrazvok ali magnetna resonanca. [ 6 ]
Genetsko ali DNK testiranje za odkrivanje najpogostejših genetskih mutacij, ki povzročajo karcinogensko melanocitno bolezen (CMT), v vzorcu krvi je omejeno, ker DNK testi trenutno niso na voljo za vse vrste bolezni. Za več informacij glejte Genetsko testiranje.
V nekaterih primerih se izvede biopsija perifernega živca (običajno suralnega živca).
Diferencialna diagnoza
Diferencialna diagnoza vključuje druge periferne nevropatije, Duchennovo mišično distrofijo, mielopatske in miastenične sindrome, diabetično nevropatijo, mielopatije pri multipli in amiotrofični lateralni sklerozi, Guillain-Barréjev sindrom, poškodbo peronealnega živca in njegovo atrofijo (vključno s stiskanjem med ledvenimi ploščicami hrbtenice), poškodbo malih možganov ali talamusa ter stranske učinke kemoterapije (med zdravljenjem s citostatiki, kot sta vinkristin ali paklitaksel). [ 7 ]
Koga se lahko obrnete?
Zdravljenje Bolezen Charcot-Marie-Tooth
Danes zdravljenje te dedne bolezni vključuje vadbeno terapijo (namenjeno krepitvi in raztezanju mišic); delovno terapijo (ki pomaga bolnikom z mišično oslabelostjo v rokah); in uporabo ortopedskih pripomočkov za lažjo hojo. Po potrebi se predpišejo protibolečinska zdravila ali antikonvulzivi. [ 8 ]
V primerih hude ploskosti stopal se lahko izvede osteotomija, v primeru deformacije pete pa je indicirana kirurška korekcija – artrodeza. [ 9 ]
Raziskave potekajo tako o genetski komponenti bolezni kot o njenih metodah zdravljenja. Uporaba matičnih celic, nekaterih hormonov, lecitina ali askorbinske kisline še ni dala pozitivnih rezultatov.
A zahvaljujoč nedavnim raziskavam se lahko v bližnji prihodnosti pri zdravljenju Charcot-Marie-Toothove bolezni resnično pojavi nekaj novega. Tako francosko podjetje Pharnext od leta 2014 razvija, od sredine leta 2019 pa potekajo klinična preskušanja zdravila PXT3003 za zdravljenje CMT tipa 1 pri odraslih, ki zavira povečano izražanje gena PMP22, izboljšuje mielinizacijo perifernih živcev in lajša nevromuskularne simptome.
Strokovnjaki iz medicinskega podjetja Sarepta Therapeutics (ZDA) delajo na ustvarjanju genske terapije za Charcot-Marie-Toothovo bolezen tipa 1. Ta terapija bo uporabljala neškodljiv adenovirus (AAV) iz rodu Dependovirus z linearnim enoverižnim genomom DNA, ki bo v telo prenesel gen NTF3, ki kodira protein nevrotrofin-3 (NT-3), potreben za delovanje Schwannovih živčnih celic.
Helixmith bo do konca leta 2020 začel s kliničnimi preskušanji genske terapije Engensis (VM202), ki jo je razvila Južna Koreja, za zdravljenje mišičnih simptomov pri karpometakarpulmonalni mikobakterijski bolezni tipa 1. [ 10 ]
Preprečevanje
Preprečevanje karcinogene melanocitne bolezni je lahko genetsko svetovanje bodočim staršem, še posebej, če ima kdo v paru družinsko anamnezo bolezni. Vendar pa so bili ugotovljeni primeri de novo točkovnih genskih mutacij, torej v odsotnosti bolezni v družinski anamnezi.
Med nosečnostjo lahko verjetnost bolezni Charcot-Marie-Tooth pri bodočem otroku preverimo z biopsijo horionskih resic (od 10. do 13. tedna nosečnosti) in z analizo amnijske tekočine (pri 15. do 18. tednu).
Napoved
Na splošno je prognoza za različne vrste Charcot-Marie-Toothove bolezni odvisna od klinične resnosti, vendar v vseh primerih bolezen napreduje počasi. Mnogi bolniki imajo invalidnost, čeprav to ne skrajša pričakovane življenjske dobe.