Medicinski strokovnjak članka

Nove publikacije
Prioni - povzročitelji prionskih bolezni
Last reviewed: 06.07.2025

Vsa vsebina iLive je pregledana ali preverjena, da se zagotovi čim večja dejanska natančnost.
Imamo stroge smernice za pridobivanje virov in samo povezave do uglednih medijskih strani, akademskih raziskovalnih institucij in, kadar je to mogoče, medicinsko pregledanih študij. Upoštevajte, da so številke v oklepajih ([1], [2] itd.) Povezave, ki jih je mogoče klikniti na te študije.
Če menite, da je katera koli naša vsebina netočna, zastarela ali drugače vprašljiva, jo izberite in pritisnite Ctrl + Enter.
Počasne virusne okužbe so značilne po posebnih merilih:
- nenavadno dolga inkubacijska doba (meseci, leta);
- specifična lezija organov in tkiv, predvsem centralnega živčnega sistema;
- počasno, enakomerno napredovanje bolezni;
- neizogiben smrtni izid.

Nekateri patogeni, ki povzročajo akutne virusne okužbe, lahko povzročijo tudi počasne virusne okužbe. Na primer, virus ošpic včasih povzroči SSPE, virus rdečk pa progresivno prirojeno rdečko in rdečki panencefalitis.
Tipično počasno virusno okužbo živali povzroča virus visna/madi, ki je retrovirus. Je povzročitelj počasne virusne okužbe in progresivne pljučnice pri ovcah. Bela možganska snov je uničena, razvije se paraliza (visna - hiranje); pojavi se kronično vnetje pljuč in vranice.
Bolezni, ki so po značilnostih podobne počasnim virusnim okužbam, povzročajo prioni - povzročitelji prionskih okužb. Prionske bolezni so skupina progresivnih motenj osrednjega živčnega sistema ljudi in živali. Pri ljudeh je delovanje osrednjega živčnega sistema oslabljeno, pride do sprememb osebnosti in motenj gibanja. Simptomi bolezni običajno trajajo od nekaj mesecev do nekaj let in se končajo s smrtjo. Prej so prionske okužbe obravnavali skupaj s tako imenovanimi povzročitelji počasnih virusnih okužb.
Nekateri povzročitelji prionskih bolezni se najprej kopičijo v limfoidnem tkivu. Prioni, ki vstopijo v možgane, se kopičijo v velikih količinah in povzročajo amiloidozo (zunajcelična disproteinoza, za katero je značilno odlaganje amiloida z razvojem atrofije in skleroze tkiva) in astrocitozo (proliferacija astrocitne nevroglije, hiperprodukcija glialnih vlaken). Nastanejo fibrili, agregati beljakovin ali amiloida in spongiformne spremembe v možganih (prenosljive spongiformne encefalopatije). Posledično se spremeni vedenje, poslabša koordinacija gibov, razvije se izčrpanost s smrtnim izidom. Imunost se ne oblikuje. Prionske bolezni so konformacijske bolezni, ki se razvijejo kot posledica nepravilnega zvijanja (kršitve pravilne konformacije) celičnih beljakovin, potrebnih za normalno delovanje telesa. Poti prenosa prionov so različne:
- prehranska pot - okuženi proizvodi živalskega izvora, aditivi za živila iz surovih govejih organov itd.:
- prenos s transfuzijo krvi, dajanjem zdravil živalskega izvora, presaditvijo organov in tkiv, uporabo okuženih kirurških in zobozdravstvenih instrumentov;
- prenos z imunobiološkimi pripravki (znana je okužba 1500 ovac s PrP''' s cepivom od možganskega formalina pri bolnih ovcah).
Patološki prioni se po vstopu v črevesje prenesejo v kri in limfo. Po periferni replikaciji v vranici, slepiču, tonzilah in drugih limfoidnih tkivih se preko perifernih živcev prenesejo v možgane (nevroinvazija). Možen je neposreden prodor prionov v možgane skozi krvno-možgansko pregrado. Prej je veljalo prepričanje, da je centralni živčni sistem edino tkivo, v katerem se kopičijo patološki prioni, vendar so se pojavile študije, ki so to hipotezo spremenile. Izkazalo se je, da je kopičenje prionov v vranici povezano s povečanjem in delovanjem folikularnih dendritičnih celic.
 [
[ Lastnosti prionov
Normalno celično izoformo prionskega proteina z molekulsko maso 33-35 kDa določa gen za prionski protein (gen za prionski protein - PrNP se nahaja na 20. človeškem kromosomu). Normalni gen se pojavi na površini celice (zasidran v membrani z glikoproteinom molekule), občutljiv na proteazo. Uravnava prenos živčnih impulzov, dnevne cikle, oksidacijske procese, sodeluje pri presnovi bakra v centralnem živčnem sistemu in pri uravnavanju delitve matičnih celic kostnega mozga. Poleg tega se prionski gen nahaja v vranici, bezgavkah, koži, prebavilih in folikularnih dendritičnih celicah.
Širjenje patoloških prionov
Do preobrazbe prionov v spremenjene oblike pride, ko je kinetično nadzorovano ravnovesje med njimi porušeno. Proces se okrepi s povečanjem količine patološkega (PrP) ali eksogenega priona. PrP je normalen protein, zasidran v celični membrani. PrP' je globularni hidrofobni protein, ki na površini celice tvori agregate s seboj in PrP'': posledično se PrP' pretvori v PrP'' in nato se cikel nadaljuje. Patološka oblika PrP''' se kopiči v nevronih, zaradi česar celica dobi gobast videz.
Kuru
Prionska bolezen, prej pogosta med Papuanci (kar pomeni tresenje ali trepetanje) v vzhodnem delu otoka Nova Gvineja. Nalezljive lastnosti bolezni je dokazal K. Gajdusek. Povzročitelj se prenaša s hrano kot posledica ritualnega kanibalizma - uživanja premalo kuhanih, s prioni okuženih možganov umrlih sorodnikov. Zaradi poškodbe osrednjega živčnega sistema sta gibanje in hoja oslabljena, pojavita se mrzlica in evforija ("smejoča se smrt"). Inkubacijska doba traja 5-30 let. Bolnik umre po enem letu.
Creutzfeldt-Jakobova bolezen
Prionska bolezen, ki se kaže kot demenca, motnje vida in možganov ter motnje gibanja s smrtnim izidom po 4-5 mesecih bolezni pri klasični varianti Creutzfeldt-Jakobove bolezni in po (3-14 mesecih) pri novi varianti Creutzfeldt-Jakobove bolezni. Inkubacijska doba lahko doseže 20 let. Možni so različni načini okužbe in vzroki bolezni:
- pri uživanju premalo toplotno obdelanih živalskih proizvodov, kot so meso in možgani krav z govejo spongiformno encefalopatijo;
- med presaditvijo tkiva, kot je presaditev roženice, transfuzija krvi, uporaba hormonov in drugih biološko aktivnih snovi živalskega izvora, uporaba katguta, kontaminiranih ali nezadostno steriliziranih kirurških instrumentov, prosekturne manipulacije;
- v primeru hiperprodukcije PrR in drugih stanj, ki spodbujajo proces pretvorbe PrR' v PrR".
Bolezen se lahko razvije tudi kot posledica mutacije ali vstavitve v regiji prionskega gena. Družinska narava bolezni je pogosta zaradi genetske predispozicije za Creutzfeldt-Jakobovo bolezen. Pri novi različici Creutzfeldt-Jakobove bolezni se motnje razvijejo v mlajši starosti (povprečna starost 28 let), v nasprotju s klasično različico (povprečna starost 65 let). Pri novi različici Creutzfeldt-Jakobove bolezni se nenormalni prionski protein kopiči ne le v osrednjem živčnem sistemu, temveč tudi v limforetikularnih tkivih, vključno s tonzilami.
Gerstmann-Sträussler-Scheinkerjev sindrom
Dedna prionska bolezen, ki jo spremljajo demenca, hipotonija, motnje požiranja (disfagija), dizartrija. Pogosto ima družinsko naravo. Inkubacijska doba je od 5 do 30 let. Bolezen se pojavi pri 50-60 letih, njeno trajanje pa se giblje od 5 do 13 let.
Dedna smrtonosna nespečnost
Avtoimunska bolezen s progresivno nespečnostjo, simpatično hiperreaktivnostjo (hipertenzija, hipertermija, hiperhidroza, tahikardija), tremorjem, ataksijo, multiklonskim sindromom, halucinacijami. Spanje je hudo moteno. Smrt nastopi s progresivno srčno-žilno odpovedjo.
Strganje
Praskavica (iz angleškega scrape - strgati) je prionska bolezen ovac in koz (scabies), ki se pojavi s poškodbo osrednjega živčnega sistema, progresivnimi motnjami gibanja, hudim srbenjem kože (scabies) in se konča s smrtjo živali.
Goveja spongiformna encefalopatija
Bolezen goveda, za katero so značilne poškodbe osrednjega živčnega sistema, motena koordinacija gibov in neizogibna smrt živali. Epidemija bolezni je prvič izbruhnila v Veliki Britaniji. Povezana je bila s hranjenjem živali z mesno-kostno moko, ki je vsebovala patološke prione. Inkubacijska doba se giblje od 1,5 do 15 let. Najbolj so okuženi možgani, hrbtenjača in zrkla živali.
Laboratorijska diagnostika prionskih bolezni
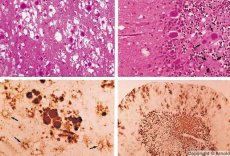
Med diagnostiko opazimo spongiformne spremembe v možganih, astrocitozo (gliozo) in odsotnost vnetnih infiltratov. Možgani se obarvajo za amiloid. V cerebrospinalni tekočini (z uporabo ELISA) se odkrijejo beljakovinski markerji prionskih možganskih motenj. Izvede se genetska analiza prionskega gena (PCR).
Preprečevanje prionskih bolezni
Za dekontaminacijo instrumentov in okoljskih predmetov se priporoča avtoklaviranje (pri 134 °C 18 min; pri 121 °C 1 h), sežig, dodatna obdelava z belilom in enonormalno raztopino NaCl 1 h. Za nespecifično profilakso so bile uvedene omejitve glede uporabe zdravil živalskega izvora, prepovedana pa je tudi proizvodnja hipofiznih hormonov živalskega izvora. Presaditev dure mater je omejena. Pri delu s tekočinami bolnikov se uporabljajo gumijaste rokavice.